Secondary Genome-Wide Association Study Using Novel Analytical Strategies Disentangle Genetic Components of Cleft Lip and/or Cleft Palate in 1q32.2


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Subjects
2.2. Conditional Genome-Wide Association Study Based on Efficient Mixed-Model Association Expedited (EMMAX) Model
2.3. Transcriptome-Wide Association Study (TWAS) Using Predicted Expression Levels
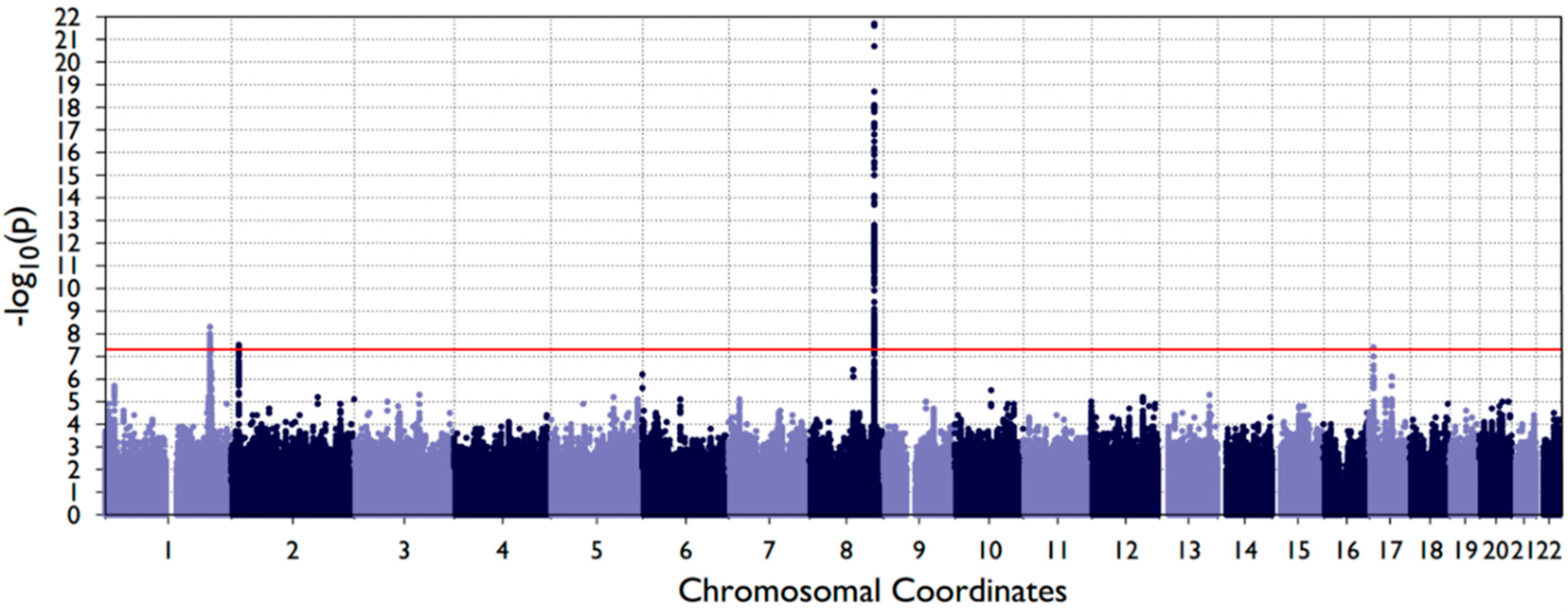
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Stanier, P.; Moore, G.E. Genetics of cleft lip and palate: Syndromic genes contribute to the incidence of non-syndromic clefts. Hum. Mol. Genet. 2004, 13, R73–R81. [Google Scholar] [CrossRef]
- Rahimov, F.; Jugessur, A.; Murray, J.C. Genetics of nonsyndromic orofacial clefts. Cleft Palate-Craniofacial J. 2011, 49, 73–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berk, N.W.; Marazita, L.M. Chapter 36. Costs of cleft lip and polate: Personal and societal implications. In Cleft Lip and Palate: From Origin to Treatment; Oxford University Press: New York, NY, USA, 2002; pp. 458–467. [Google Scholar]
- Grosen, D.; Chevrier, C.; Skytthe, A.; Bille, C.; Mølsted, K.; Sivertsen, Å.; Murray, J.C.; Christensen, K. A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: Support for the multifactorial threshold model of inheritance. J. Med. Genet. 2009, 47, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Sivertsen, Å.; Wilcox, A.J.; Skjaerven, R.; Vindenes, H.A.; Åbyholm, F.; Harville, E.; Lie, R.T. Familial risk of oral clefts by morphological type and severity: Population based cohort study of first degree relatives. BMJ 2008, 336, 432–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosen, D.; Bille, C.; Petersen, I.; Skytthe, A.; Hjelmborg, J.V.B.; Pedersen, J.K.; Murray, J.C.; Christensen, K. Risk of Oral Clefts in Twins. Epidemiology 2011, 22, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Marazita, M. Chapter 18. Segregation Analyses. In Cleft Lip and Palate: From Origin to Treatment; Oxford University Press: New York, NY, USA, 2002; pp. 222–233. [Google Scholar]
- Marazita, M.L.; Murray, J.C.; Lidral, A.C.; Arcos-Burgos, M.; Cooper, M.E.; Goldstein, T.; Maher, B.S.; Daack-Hirsch, S.; Schultz, R.; Mansilla, M.A.; et al. Meta-Analysis of 13 Genome Scans Reveals Multiple Cleft Lip/Palate Genes with Novel Loci on 9q21 and 2q32-35. Am. J. Hum. Genet. 2004, 75, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marazita, M.L.; Lidral, A.C.; Murray, J.C.; Field, L.; Maher, B.S.; McHenry, T.G.; Cooper, M.E.; Govil, M.; Daack-Hirsch, S.; Riley, B.; et al. Genome Scan, Fine-Mapping, and Candidate Gene Analysis of Non-Syndromic Cleft Lip with or without Cleft Palate Reveals Phenotype-Specific Differences in Linkage and Association Results. Hum. Hered. 2009, 68, 151–170. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, K.U.; Böhmer, A.C.; Rubini, M.; Mossey, P.A.; Herms, S.; Nowak, S.; Reutter, H.; Alblas, M.A.; Lippke, B.; Barth, S.; et al. Strong Association of Variants around FOXE1 and Orofacial Clefting. J. Dent. Res. 2014, 93, 376–381. [Google Scholar] [CrossRef]
- Birnbaum, S.; Ludwig, K.U.; Reutter, H.M.; Herns, S.; Steffens, M.; Rubini, M.; Baluardo, C.; Ferrian, M.; De Assis, N.A.; Alblas, M.A.; et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 2009, 41, 473–477. [Google Scholar] [CrossRef]
- Grant, S.F.; Wang, K.; Zhang, H.; Glaberson, W.; Annaiah, K.; Kim, C.E.; Bradfield, J.P.; Glessner, J.T.; Thomas, K.A.; Garris, M.; et al. A Genome-Wide Association Study Identifies a Locus for Nonsyndromic Cleft Lip with or without Cleft Palate on 8q24. J. Pediatr. 2009, 155, 909–913. [Google Scholar] [CrossRef]
- Beaty, T.H.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Ruczinski, I.; Hetmanski, J.B.; Liang, K.Y.; Wu, T.; Murray, T.; Fallin, M.D.; et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 2010, 42, 525–529. [Google Scholar] [CrossRef]
- Mangold, E.; Ludwig, K.U.; Birnbaum, S.; Baluardo, C.; Ferrian, M.; Herms, S.; Reutter, H.M.; De Assis, N.A.; Al Chawa, T.; Mattheisen, M.; et al. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat. Genet. 2009, 42, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, K.U.; Mangold, E.; Herms, S.; Nowak, S.; Reutter, H.; Paul, A.; Becker, J.; Herberz, R.; AlChawa, T.; Nasser, E.; et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 2012, 44, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaty, T.H.; Taub, M.A.; Scott, A.F.; Murray, J.C.; Marazita, M.L.; Schwender, H.; Parker, M.; Hetmanski, J.B.; Balakrishnan, P.; Mansilla, M.A.; et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum. Genet. 2013, 132, 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Huang, Y.; Yin, A.; Pan, Y.; Wang, Y.; Wang, C.; Du, Y.; Wang, M.; Lan, F.; Hu, Z.; et al. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat. Commun. 2015, 6, 6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, E.J.; Liu, H.; Carlson, J.C.; Shaffer, J.R.; Feingold, E.; Wehby, G.; Laurie, C.A.; Jain, D.; Laurie, C.C.; Doheny, K.F.; et al. A Genome-wide Association Study of Nonsyndromic Cleft Palate Identifies an Etiologic Missense Variant in GRHL3. Am. J. Hum. Genet. 2016, 98, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Leslie, E.J.; Carlson, J.C.; Shaffer, J.R.; Feingold, E.; Wehby, G.; Laurie, C.A.; Jain, D.; Laurie, C.C.; Doheny, K.F.; McHenry, T.; et al. A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13. Hum. Mol. Genet. 2016, 25, 2862–2872. [Google Scholar] [CrossRef] [Green Version]
- Mangold, E.; Böhmer, A.C.; Ishorst, N.; Hoebel, A.-K.; Gültepe, P.; Schuenke, H.; Klamt, J.; Hofmann, A.; Gölz, L.; Raff, R.; et al. Sequencing the GRHL3 Coding Region Reveals Rare Truncating Mutations and a Common Susceptibility Variant for Nonsyndromic Cleft Palate. Am. J. Hum. Genet. 2016, 98, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Leslie, E.J.; Carlson, J.C.; Shaffer, J.R.; Buxó, C.J.; Castilla, E.E.; Christensen, K.; Deleyiannis, F.W.B.; Field, L.L.; Hecht, J.T.; Moreno, L.; et al. Association studies of low-frequency coding variants in nonsyndromic cleft lip with or without cleft palate. Am. J. Med Genet. Part A 2017, 173, 1531–1538. [Google Scholar] [CrossRef]
- Leslie, E.J.; Carlson, J.C.; Shaffer, J.R.; Butali, A.; Buxó, C.J.; Castilla, E.E.; Christensen, K.; Deleyiannis, F.W.B.; Field, L.L.; Hecht, J.T.; et al. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum. Genet. 2017, 136, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zuo, X.; He, M.; Gao, J.; Fu, Y.; Qin, C.; Meng, L.; Wang, W.; Song, Y.; Cheng, Y.; et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 2017, 8, 14364. [Google Scholar] [CrossRef] [PubMed]
- Eshete, M.; Liu, H.; Li, M.; Adeyemo, W.; Gowans, L.; Mossey, P.; Busch, T.; Deressa, W.; Donkor, P.; Olaitan, P.; et al. Loss-of-Function GRHL3 Variants Detected in African Patients with Isolated Cleft Palate. J. Dent. Res. 2017, 97, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Beaty, T.H.; Ruczinski, I.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Hetmanski, J.B.; Murray, T.; Redett, R.J.; Fallin, M.D.; Liang, K.Y.; et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet. Epidemiol. 2011, 35, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Haaland, Ø.A.; Lie, R.T.; Romanowska, J.; Gjerdevik, M.; Gjessing, H.K.; Jugessur, A. A Genome-Wide Search for Gene-Environment Effects in Isolated Cleft Lip with or without Cleft Palate Triads Points to an Interaction between Maternal Periconceptional Vitamin Use and Variants in ESRRG. Front. Genet. 2018, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Curà, F.; Palmieri, A.; Girardi, A.; Carinci, F.; Morselli, P.G.; Nouri, N.; Pezzetti, F.; Scapoli, L.; Martinelli, M. Possible effect of SNAIL family transcriptional repressor 1 polymorphisms in non-syndromic cleft lip with or without cleft palate. Clin. Oral Investig. 2018, 22, 2535–2541. [Google Scholar] [CrossRef]
- Estandia-Ortega, B.; Velázquez-Aragón, J.A.; Alcántara-Ortigoza, M.A.; Reyna-Fabián, M.E.; Villagómez-Martínez, S.; Angel, A.G.-D. 5,10-Methylenetetrahydrofolate reductase single nucleotide polymorphisms and gene-environment interaction analysis in non-syndromic cleft lip/palate. Eur. J. Oral Sci. 2014, 122, 109–113. [Google Scholar] [CrossRef]
- Wu, T.; Fallin, M.D.; Shi, M.; Ruczinski, I.; Liang, K.Y.; Hetmanski, J.B.; Wang, H.; Ingersoll, R.G.; Huang, S.; Ye, X.; et al. Evidence of gene-environment interaction for the RUNX2 gene and environmental tobacco smoke in controlling the risk of cleft lip with/without cleft palate. Birth Defects Res. Part A Clin. Mol. Teratol. 2012, 94, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Niktabar, S.M.; Aarafi, H.; Dastgheib, S.A.; NooriShadkam, M.; Mirjalili, S.R.; Lookzadeh, M.H.; Akbarian-Bafghi, M.J.; Morovati-Sharifabad, M.; Neamatzadeh, H. Association of MTHFR 1298A > C Polymorphism with Susceptibility to Non-Syndromic Cleft Lip with or without Palate: A Case-Control Study and Meta-Analysis. Fetal Pediatr. Pathol. 2019, 1–17. [Google Scholar] [CrossRef]
- Li, W.; Wang, M.; Zhou, R.; Wang, S.; Zheng, H.; Liu, D.; Zhou, Z.; Zhu, H.; Wu, T.; Beaty, T.H. Exploring the interaction between FGF Genes and T-box genes among chinese nonsyndromic cleft lip with or without cleft palate case-parent trios. Environ. Mol. Mutagen. 2019, 60, 602–606. [Google Scholar] [CrossRef]
- Jugessur, A.; Rahimov, F.; Lie, R.T.; Wilcox, A.J.; Gjessing, H.K.; Nilsen, R.M.; Nguyen, T.T.; Murray, J.C. Genetic variants inIRF6and the risk of facial clefts: Single-marker and haplotype-based analyses in a population-based case-control study of facial clefts in Norway. Genet. Epidemiol. 2008, 32, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Rahimov, F.; Program, N.C.S.; Marazita, M.L.; Visel, A.; Cooper, M.E.; Hitchler, M.J.; Rubini, M.; Domann, F.E.; Govil, M.; Christensen, K.; et al. Disruption of an AP-2α binding site in an IRF6 enhancer is associated with cleft lip. Nat. Genet. 2008, 40, 1341–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingraham, C.R.; Kinoshita, A.; Kondo, S.; Yang, B.; Sajan, S.; Trout, K.J.; Malik, M.I.; Dunnwald, M.; Goudy, S.L.; Lovett, M.; et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 2006, 38, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Mendoza, F.; Tan, E.; Bertol, J.W.; Gaggar, A.S.; Jun, G.; Biguetti, C.; Fakhouri, W.D. A cleft lip and palate gene, Irf6, is involved in osteoblast differentiation of craniofacial bone. Dev. Dyn. 2019, 248, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Leslie, E.J.; Taub, M.A.; Liu, H.; Steinberg, K.M.; Koboldt, D.C.; Zhang, Q.; Carlson, J.C.; Hetmanski, J.B.; Wang, H.; Larson, D.E.; et al. Identification of Functional Variants for Cleft Lip with or without Cleft Palate in or near PAX7, FGFR2, and NOG by Targeted Sequencing of GWAS Loci. Am. J. Hum. Genet. 2015, 96, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G. GTEx detects genetic effects. Science 2015, 348, 640–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.L.; Cox, N.J.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ehuppi, K.; Pitt, J.J.; Wahlberg, B.M.; Caplen, N.J. The 8q24 Gene Desert: An Oasis of Non-Coding Transcriptional Activity. Front. Genet. 2012, 3, 69. [Google Scholar] [CrossRef] [Green Version]
- Glass, D.N.; Giannini, E.H. Juvenile rheumatoid arthritis as a complex genetic trait. Arthritis Rheum. 1999, 42, 2261–2268. [Google Scholar] [CrossRef]
- Haiman, C.A.; Patel, Y.M.; Stram, D.O.; Carmella, S.G.; Chen, M.; Wilkens, L.R.; Le Marchand, L.; Hecht, S.S. Benzene Uptake and Glutathione S-transferase T1 Status as Determinants of S-Phenylmercapturic Acid in Cigarette Smokers in the Multiethnic Cohort. PLoS ONE 2016, 11, e0150641. [Google Scholar] [CrossRef] [Green Version]
- Koettgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2012, 45, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Beaty, T.H.; Marazita, M.L.; Leslie, E.J. Genetic factors influencing risk to orofacial clefts: Today’s challenges and tomorrow’s opportunities. F1000Research 2016, 5, 2800. [Google Scholar] [CrossRef]
- Zheng, X.; Feingold, E.; Ryckman, K.K.; Shaffer, J.R.; Boyd, H.A.; Feenstra, B.; Melbye, M.; Marazita, M.L.; Murray, J.C.; Cuenco, K.T. Association of maternal CNVs in GSTT1/GSTT2 with smoking, preterm delivery, and low birth weight. Front. Genet. 2013, 4, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammer, E.J.; Shaw, G.M.; Iovannisci, D.M.; Finnell, R.H. Maternal Smoking, Genetic Variation of Glutathione S-Transferases, and Risk for Orofacial Clefts. Epidemiology 2005, 16, 698–701. [Google Scholar] [CrossRef]
- Hozyasz, K.K.; Mostowska, A.; Surowiec, Z.; Jagodziński, P.P. Genetic polymorphisms of GSTM1 and GSTT1 in mothers of children with isolated cleft lip with or without cleft palate. Przeglad Lek. 2005, 62, 1019–1022. [Google Scholar]
- Shi, M.; Christensen, K.; Weinberg, C.R.; Romitti, P.; Bathum, L.; Lozada, A.; Morris, R.W.; Lovett, M.; Murray, J.C. Orofacial Cleft Risk Is Increased with Maternal Smoking and Specific Detoxification-Gene Variants. Am. J. Hum. Genet. 2007, 80, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Kitani-Morii, F.; Imamura, K.; Kondo, T.; Ohara, R.; Enami, T.; Shibukawa, R.; Yamamoto, T.; Sekiguchi, K.; Toguchida, J.; Mizuno, T.; et al. Analysis of neural crest cells from Charcot–Marie–Tooth disease patients demonstrates disease-relevant molecular signature. NeuroReport 2017, 28, 814–821. [Google Scholar] [CrossRef]
- Bolt, H.M.; Thier, R. Relevance of the Deletion Polymorphisms of the Glutathione S-Transferases GSTT1 and GSTM1 in Pharmacology and Toxicology. Curr. Drug Metab. 2006, 7, 613–628. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J.; Choi, J. γ-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhang, W.; Sehmi, J.; Li, X.; Wass, M.N.; Van Der Harst, P.; Holm, H.; Sanna, S.; Kavousi, M.; Baumeister, S.E.; et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011, 43, 1131–1138. [Google Scholar] [CrossRef]
- Mishra, S.; Sabhlok, S.; Panda, P.K.; Khatri, I. Management of Midline Facial Clefts. J. Maxillofac. Oral Surg. 2015, 14, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Sebo, Z.L.; Jeffery, E.; Holtrup, B.; Rodeheffer, M.S. A mesodermal fate map for adipose tissue. Development 2018, 145, dev166801. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Characteristics | Categories | Control (n = 7839) | Case (n = 2703) |
|---|---|---|---|
| Cleft Type | Cleft Lip only | - | 620 |
| Cleft Lip and Palate | - | 1771 | |
| Cleft Palate only | - | 312 | |
| Sex | Male | 3397 (43.3%) | 1581 (58.50%) |
| Female | 4442 (56.7%) | 1122 (41.50%) | |
| Age | 28.66 (±16.95) | 12.06 (±13.03) | |
| Race | African | 219 (2.79%) | 160 (5.92%) |
| Asian | 1433 (18.28%) | 584 (21.60%) | |
| Caucasian | 3563 (45.45%) | 1068 (39.51%) | |
| Caucasian and African | 33 (0.42%) | 11 (0.41%) | |
| Caucasian and Asian | 2 (0.026%) | 2 (0.074%) | |
| Caucasian and Native American | 768 (9.80%) | 171 (6.33%) | |
| Caucasian, African and Native American | 1617 (20.63%) | 665 (24.60%) | |
| Native American | 152 (1.94%) | 37 (1.37%) | |
| Unspecified | 52 (0.66%) | 5 (0.18%) | |
| Locus | EMMAX Analysis | The Multiethnic Meta-Analysis [19] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sentinel SNP | Chr:Pos | Ref/Alt | MAF | β (S.E.) | p-Value | Gene | Sentinel SNP | Meta OR † | p-Value | r2 ‡ | |
| 8q24 | rs72728755 | 8:129990382 | T/A | 0.16 | 0.08 (0.008) | 1.94 × 10−22 | Gene desert | rs17242358 | 2.00 [1.78–2.26] | 5.63 × 10−30 | 0.97 |
| 1q32.2 | rs2235370 | 1:209946027 | C/A | 0.13 | −0.05 (0.009) | 5.15 × 10−9 | TRAF3IP3 | rs11119345 | 1.81 [1.57–2.07] | 2.52 × 10−17 | 0.84 |
| rs2235373 | 1:209963803 | G/A | 0.26 | −0.04 (0.007) | 2.12 × 10−8 | IRF6 | 0.30 | ||||
| 2p24.2 | rs7552 | 2:16733928 | A/G | 0.50 | 0.04 (0.006) | 3.40 × 10−8 | FAM49A | rs7552 | 1.28 [1.17–1.40] | 4.22 × 10−8 | 1.00 |
| 17p13.1 | rs11273201 | 17:8930219 | AACCCAAAACCCAC/A | 0.27 | −0.04 (0.007) | 3.58 × 10−8 | NTN1 | rs11273201 | 1.44 [1.29–1.59] | 7.84 × 10−12 | 1.00 |
| No. | SNP ID | Chr:Pos | Ref/Alt | MAF | β (S.E.) | p-Value | Locus/Gene |
|---|---|---|---|---|---|---|---|
| 1 | rs72728755 | 8:129990382 | T/A | 0.16 | 0.08 (0.008) | 1.94 × 10−22 | 8q24 |
| 2 | rs7552 | 2:16733928 | A/G | 0.50 | 0.03 (0.006) | 2.67 × 10−8 | FAM49A |
| 3 | rs2235370 | 1:209946027 | C/A | 0.13 | −0.05 (0.009) | 1.33 × 10−8 | TRAF3IP3 |
| 4 | rs16957821 | 17:8948104 | C/G | 0.24 | 0.04 (0.007) | 1.40 × 10−7 | NTN1 |
| 5 | rs12600562 | 17:44977040 | G/T | 0.47 | −0.03 (0.006) | 7.06 × 10−7 | LRRC37A2 |
| 6 | rs12543318 | 8:88868340 | C.A | 0.45 | −0.03 (0.006) | 5.43 × 10−7 | 8q21 |
| 7 | rs3845903 | 3:66517670 | C/T | 0.03 | −0.10 (0.020) | 2.79 × 10−6 | LRIG1 |
| 8 | rs10778143 | 12:102094643 | A/C | 0.33 | 0.03 (0.007) | 1.76 × 10−6 | CHPT1 |
| 9 | rs6929507 | 6:1632072 | T/C | 0.01 | 0.13 (0.027) | 2.14 × 10−6 | GMDS |
| 10 | rs138322543 | 9:100641771 | CCACCA/C | 0.27 | 0.03 (0.007) | 4.82 × 10−6 | TRMO |
| Gene | Ensembl ID | GRCh37 Coordinate | β (S.E.) | z | p-Value |
|---|---|---|---|---|---|
| Whole blood | |||||
| THEM79 | ENSG00000163472 | 1: 156,252,726–156,262,976 | 2.71 (0.56) | 4.87 | 1.13 × 10−6 |
| GLMP | ENSG00000198715 | 1: 156,259,880–156,265,480 | 1.57 (0.33) | 4.83 | 1.37 × 10−6 |
| Skeletal muscle | |||||
| DDT | ENSG00000099977 | 22: 24,313,554–24,322,660 | 1.12 (0.21) | 5.25 | 1.52 × 10−7 |
| Adipose (Subcutaneous) | |||||
| MIF | ENSG00000240972 | 22: 24,236,191–24,237,414 | 1.79 (0.28) | 6.39 | 1.62 × 10−10 |
| MIF-AS1 | ENSG00000218537 | 22: 24,236,613–24,241,117 | 1.39 (0.24) | 5.87 | 4.25 × 10−9 |
| DDT | ENSG00000099977 | 22: 24,313,554–24,322,660 | 2.94 (0.54) | 5.43 | 5.66 × 10−8 |
| GSTT2 | ENSG00000099984 | 22: 24,322,249–24,326,106 | −1.42 (0.28) | −5.16 | 2.46 × 10−7 |
| USP2 | ENSG00000036672 | 11: 119,225,925–119,252,436 | −1.75 (0.38) | −4.57 | 4.93 × 10−6 |
| DDTL | ENSG00000099974 | 22: 24,309,089–24,314,721 | −1.46 (0.32) | −4.54 | 5.68 × 10−6 |
| Adipose (Visceral) | |||||
| MIF | ENSG00000240972 | 22: 24,236,191–24,237,414 | 1.66 (0.26) | 6.52 | 7.09 × 10−11 |
| DDTL | ENSG00000099974 | 22: 24,309,089–24,314,721 | −2.60 (0.45) | −5.81 | 6.37 × 10−9 |
| MIF-AS1 | ENSG00000218537 | 22: 24,236,613–24,241,117 | 1.10 (0.19) | 5.78 | 7.31 × 10−9 |
| GSTT2 | ENSG00000099984 | 22: 24,322,249–24,326,106 | −1.05 (0.19) | −5.53 | 3.27 × 10−8 |
| GSTT2B | ENSG00000133433 | 22: 24,299,601–24,303,373 | −1.36 (0.28) | −4.94 | 7.92 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Suzuki, A.; Iwata, J.; Jun, G. Secondary Genome-Wide Association Study Using Novel Analytical Strategies Disentangle Genetic Components of Cleft Lip and/or Cleft Palate in 1q32.2. Genes 2020, 11, 1280. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111280
Yang Y, Suzuki A, Iwata J, Jun G. Secondary Genome-Wide Association Study Using Novel Analytical Strategies Disentangle Genetic Components of Cleft Lip and/or Cleft Palate in 1q32.2. Genes. 2020; 11(11):1280. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111280
Chicago/Turabian StyleYang, Yunju, Akiko Suzuki, Junichi Iwata, and Goo Jun. 2020. "Secondary Genome-Wide Association Study Using Novel Analytical Strategies Disentangle Genetic Components of Cleft Lip and/or Cleft Palate in 1q32.2" Genes 11, no. 11: 1280. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111280