Genetic Mutations and Variants in the Susceptibility of Familial Non-Medullary Thyroid Cancer
by
 ,
,
Fabíola Yukiko Miasaki
1 ,
,
Cesar Seigi Fuziwara
2,
Gisah Amaral de Carvalho
1 and
Edna Teruko Kimura
2,* 1
Department of Endocrinology and Metabolism (SEMPR), Hospital de Clínicas, Federal University of Paraná, Curitiba 80030-110, Brazil
2
Department of Cell and Developmental Biology, Institute of Biomedical Sciences, University of São Paulo, São Paulo 05508-000, Brazil
*
Author to whom correspondence should be addressed.
Genes 2020, 11(11), 1364; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111364
Submission received: 24 October 2020
/
Revised: 11 November 2020
/
Accepted: 16 November 2020
/
Published: 18 November 2020
(This article belongs to the Special Issue Genetic Perspectives in Thyroid Cancer)
Abstract
:Thyroid cancer is the most frequent endocrine malignancy with the majority of cases derived from thyroid follicular cells and caused by sporadic mutations. However, when at least two or more first degree relatives present thyroid cancer, it is classified as familial non-medullary thyroid cancer (FNMTC) that may comprise 3–9% of all thyroid cancer. In this context, 5% of FNMTC are related to hereditary syndromes such as Cowden and Werner Syndromes, displaying specific genetic predisposition factors. On the other hand, the other 95% of cases are classified as non-syndromic FNMTC. Over the last 20 years, several candidate genes emerged in different studies of families worldwide. Nevertheless, the identification of a prevalent polymorphism or germinative mutation has not progressed in FNMTC. In this work, an overview of genetic alteration related to syndromic and non-syndromic FNMTC is presented.
1. Introduction
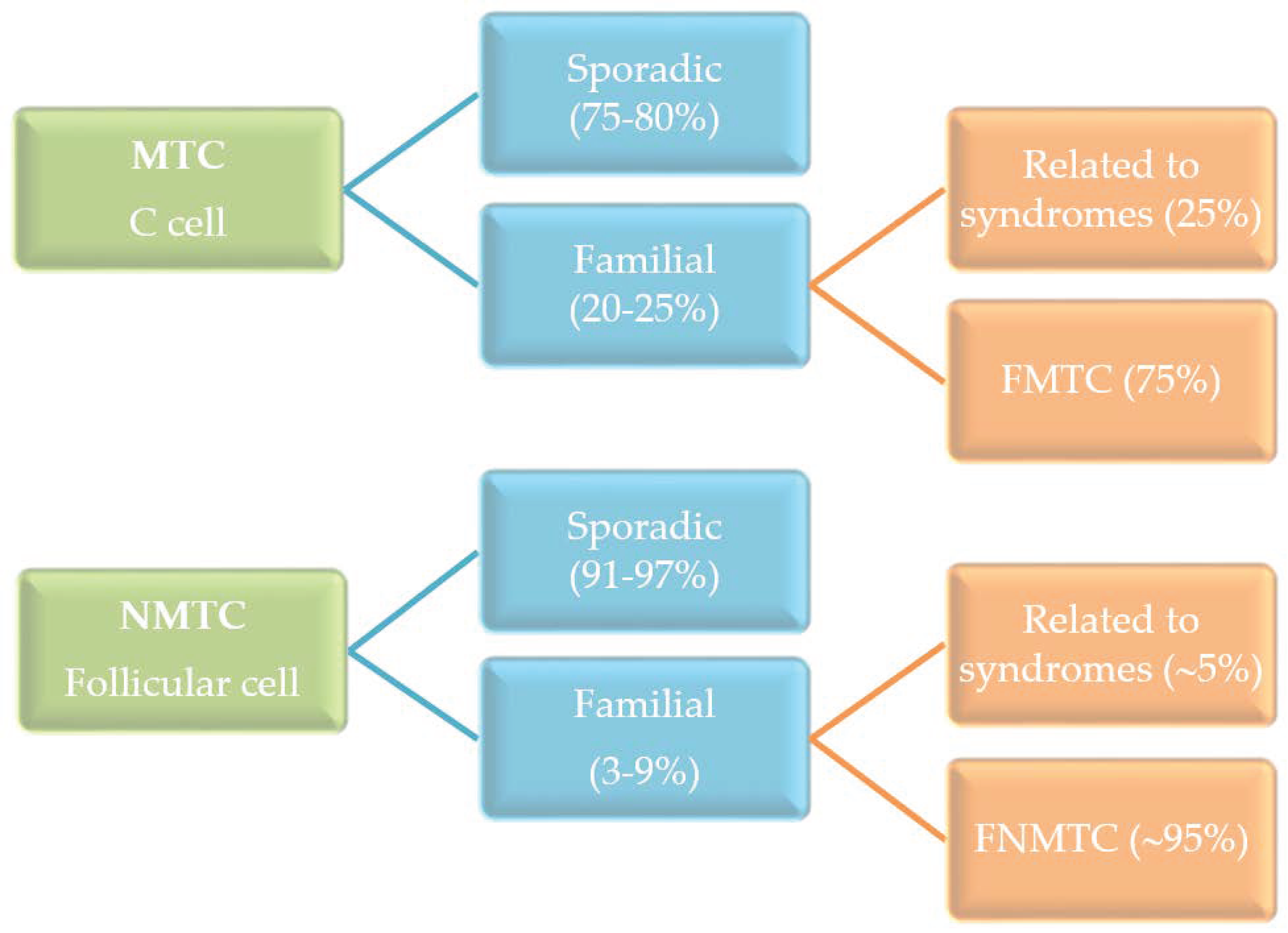
The most common type of thyroid cancer derives from thyroid follicular cells and is named as non-medullary thyroid cancer (NMTC) in order to be distinguished from the less frequent medullary thyroid cancer (MTC) that originates from the thyroid C-cells. The MTC occurs as sporadic and hereditary cancer, in contrast to the NMTC, which is mainly sporadic (Figure 1). The hereditary MTC can be a component of a syndrome or have a familial background. In this context, the NMTC can also be associated with syndromic conditions, such as in Cowden syndrome, Carney complex, Werner syndrome, and familial adenomatous polyposis but to a lesser extent than in MTC. Moreover, a high prevalence of NMTC in ataxia-telangiectasia, DICER1, and Pendred syndromes has been described [1,2].
Besides these well-known genetic syndromes, the characterization of the non-syndromic form of familial non-medullary thyroid cancer (FNMTC) remains to be consolidated. In 1953, Firminger and Skelton reported the first case of papillary thyroid cancer (PTC) in twins [3]. However, the concept of FNMTC and the genetic predisposition to PTC has emerged only in recent decades. Currently, it is accepted that FNMTC occurs when two or more first-degree relatives are diagnosed with NMTC cancer [4].
The initial FNMTC studies were performed by linkage analysis and described some specific loci, although they did not identify a precise gene associated with FMNTC [5,6,7,8,9,10]. Furthermore, despite the efforts of many groups in investigating FNMTC using Sanger sequencing, no conclusive information was found, suggesting genetic heterogeneity, multigenic inheritance, and multifactorial inheritance [11]. However, a new genomic perspective emerged with the application of Next Generation Sequencing (NGS) technology that covered the entire genome. In this extent, some new insights into genetics of FNMTC have emerged by the recent genome-wide association studies (GWAS) in populations of PTC. The finding of several single nucleotide polymorphisms (SNPs), such as in DIRC3, NIRG1, FOXE1, NKX2-1, and PCNXL2, were observed in the European, Korean, and American populations [12]. Increasing evidence suggests that genetic predisposition factors play an essential role in carcinogenesis besides environmental factors [13]. In this review, we cover the genetic findings associated with FNMTC and in syndromes related to NMTC.
2. Syndromic Causes of Non-Medullary Thyroid Cancer
Many syndromes associated with thyroid tumor predisposition have Mendelian patterns of inheritance, and they are related to mutations that may influence the mechanism of DNA repair, the microRNA processing, and maturation, the genome integrity maintenance, the cell signaling, or mitochondrial regulated cellular processes (Table 1) [13]. These syndromes are characterized by several other main malignancies and sometimes lack to present thyroid cancer. Some authors have suggested surveillance in syndromic FNMTC (see below). However, the guidelines, such as the 2015 American Thyroid Association’s guideline, are precautious due to insufficient evidence in order to recommend the thyroid cancer screening [14].
2.1. Cowden Syndrome
Cowden syndrome (OMIM #158350) is characterized by hamartomas in different parts of the body (gastrointestinal hamartomas, ganglioneuromas, trichilemmomas) associated with melanomas, breast, endometrial and thyroid cancer, macrocephaly, and, eventually, autism spectrum disorder and/or mental retardation [18,28]. One of the major diagnosis criteria is the presence of follicular thyroid carcinoma (FTC). However, if it is not detected, a biannual thyroid ultrasound is advocated in patients older than seven years [29].
Cowden syndrome is classically associated with mutations in the phosphatase and tensin homolog (PTEN) gene on chromosome 10q22–23, although variants in several other genes have been described in patients without PTEN mutations (SDHB-D, SEC23B, KLLN, PARP4, AKT1, PIK3CA, USF3, TTN, MUTYH, RET, TSC2, BRCA1, BRCA2, ERCC2, HRAS, and RASAL1) [19,30,31,32].
PTEN dephosphorylates PI (3,4,5) P3 (PIP3) and PI (3, 4) P2 to PI (4,5) P2 and PI (4) P, respectively. Thus, PIP3 and PI (3,4) P2 do not activate AKT serine/threonine kinase (AKT), and the phosphatidilinositol 3-kinase (PI3K)/AKT signaling pathway remains inhibited. PTEN mutation results in loss of function, leading to a high concentration of PI (3,4) P2 that activates AKT and enhances cell proliferation, cell migration, and reduces cell death [33,34]. In addition, mechanisms that regulate PTEN expression and compartmentalization are involved in tumorigenesis [35].
The first correlations between the PIK3-AKT pathway activation and thyroid cancer were observed in Cowden syndrome studies. Since Cowden syndrome mainly presents with FTC and PTEN activates the PIK3-AKT pathway, some authors have postulated that PIK3-AKT activation is required for FTC oncogenesis, and these preliminary findings were further corroborated [36,37].
Another intriguing fact was the association of RAS protein activator like 1 (RASAL1) with Cowden Syndrome. RASAL1 is a negative modulator of the RAS signaling pathway and suppresses both mitogen-activated protein kinase (MAPK) and PI3K pathways. However, RASAL1 is frequently found methylated or mutated in sporadic follicular and anaplastic thyroid cancer [38].
In a large series of 155 patients with Cowden syndrome and thyroid cancer, 39 presented with PTEN germline mutations, while RASAL1 germline alteration (RASAL1, c.982C>T, R328W) was observed in two patients without PTEN mutations [32]. In the same study, the authors also analyzed the germline database of The Cancer Genome Atlas (TCGA) and discovered that 0.6% of PTC patients harbored the deleterious germline RASAL1 mutation [32].
2.2. Carney Complex
The Carney complex (OMIM #160980) is an autosomal dominant disorder in 70% of the cases, characterized by loss-of-function mutations in the PRKAR1A gene (17q22–24).
Under a normal condition of several endocrine related ligands, such as TSH, FSH, ACTH, GHRH, and MSH, when binding to the G-protein coupled receptor activates protein kinase A (PKA). PRKAR1A encodes the R1α subunit of PKA. Thus, when mutated, it increases cAMP-dependent PKA activity and drives tumorigenesis [17,39,40]. Therefore, thyrocytes, Sertoli cells, adrenocortical cells, somatotrophs, and melanocytes are directly affected by the PRKAR1a mutation. As a result, variable endocrine tumors are observed in the Carney complex disease, including primary pigmented nodular adrenocortical disease, pituitary adenomas, testicular tumors, ovarian lesions, and myxomas and lentiginosis syndromes [17]. Since thyroid cancer could also be part of this syndrome, annual long-term surveillance is recommended [17].
Evidence shows that PRKAR1A acts as a tumor suppressor gene in sporadic thyroid cancers [41]. However, the traditional thyroid cancer pathways (MAPK and PIK3-AKT pathways) are not involved in the Carney complex [42]. Instead, a recent in vitro study suggests that PKA activates AMP-activated kinase (AMPK) through serine/threonine kinase 11 (LKB1, also named SKT11) in Carney-related FTC without inhibiting mTOR activation [43].
2.3. Werner Syndrome
Werner syndrome is one of the progeroid syndromes (OMIM #27770) characterized by early aging, scleroderma-like skin changes, bilateral cataracts, and subcutaneous calcifications, premature arteriosclerosis, and diabetes mellitus. Different types of cancers are associated with this syndrome, such as meningiomas, myeloid disorders, soft tissue sarcomas, and thyroid carcinoma [1,44]. Their regular surveillance is recommended [45]. The Werner Syndrome’s patients carry autosomal recessive WRN RecQ like helicase (WRN) gene mutations on 8p11.1–21.1. WRN gene encodes RecQ helicase that regulate DNA replication, recombination, repair, transcription, and telomerase maintenance. Dysregulation of this pathway triggers DNA instability, telomeric fusions of homologous chromosomes, and, ultimately, oncogenesis [13]. However, the precise mechanisms that contribute to genome instability in Werner syndrome remains unclear [46]. In a Japanese series, mutations in the N-terminal portion of WRN was correlated with PTC, while mutations in C-terminal with FTC [47]. The N-terminal portion of WRN contains exonuclease activity, whereas the central part contains the DNA-dependent ATPase, 3′–5′ helicase, and annealing activity [46]. Overall, these studies suggest specific effects in WRN activity depending on the site of mutation. Moreover, an in vitro study showed that mutations in WRN’s nuclease domain, helicase domain, or DNA binding domain aborted its canonical stimulatory effect on nonhomologous end-joining (c-NHEJ) pathway during DNA double-strand break (DSB) repair [46].
2.4. Familial Adenomatous Polyposis
The phenotype of familial adenomatous polyposis (FAP) (OMIM #175100) is characterized by numerous intestinal polyps, colon cancer, and other cancers that include thyroid cancer [2,22,48]. FAP is an autosomal dominant disorder caused by mutations in APC regulator of WNT signaling pathway (APC) gene on chromosome 5q21. The APC gene is a suppressor of the Wnt signaling pathway and regulates β-catenin activation by multiple mechanisms. In normal conditions, the Axin complex—formed by APC, glycogen synthase kinase 3 (GSK3), and casein kinase 1 (CK1)—phosphorylates the amino-terminal of the free β-catenin, permitting its recognition and further ubiquitination [49,50]. By this process of continuous degradation, β-catenin remains in the cytoplasm without reaching the promoter region of target genes in the nucleus. Thus, when the APC protein is mutated or truncated, β-catenin is released from its degradation and migrates to the nucleus, activating gene transcription of oncogenic pathways. Truncated APC protein also interferes with chromosome stability and cell migration [50].
In addition to the germline mutation, biallelic inactivation of the wild-type APC allele is frequently necessary for tumorigenesis, and the second-hit is commonly acquired by somatic mutation [51]. In the FAP-associated thyroid cancer, the concomitant presence of germline and distinct somatic mutation were observed in several Japanese families [48,52,53]. Most of FAP-associated thyroid cancers present the histological subtype called cribriform-morular variant of PTC (CMVPTC) [2,51]. An annual thyroid ultrasound is recommended to late teen years’ patients [54,55].
2.5. Ataxia-Telangiectasia Syndrome
Ataxia-telangiectasia (A-T) syndrome (OMIM #208900) is an autosomal recessive disorder linked to the mutation of the ATM serine/threonine kinase (ATM) gene and characterized by degenerative cerebellar atrophy, telangiectasias, immune defects, and malignancy [56,57]. It is also well-known that relatives of patients with ataxia-telangiectasia have an increased cancer incidence [16].
ATM protein belongs to the PI-3 kinase-like protein kinases family. Besides TP53, BRCA1, and BRCA2, ATM is considered a genome’s guardian and participates directly in the DNA damage response (DDR). For its activation, MRE11-RAD50-NBS1 (MRN) complex—A sensor of DSB (double strand-break)—induces several autophosphorylations and acetylations. Activated ATM then phosphorylates different proteins involved in the DSB (double-strand break) response [58]. For instance, ATM phosphorylates CHK2 and p53, which are both involved in senescence and apoptosis [58].
An increased incidence of thyroid cancer was observed in obligate ATM mutation carriers (RR adjusted = 2.6) [16]. Later, selective mutations in the ATM gene are related to thyroid cancer. ATM c.2119T>C p.S707P (rs4986761) heterozygotes were associated with an adjusted HR (hazard ratio for cancer) of 10 for thyroid/endocrine tumors, while no association was observed in ATM c.146C>G p.S49C (rs1800054) heterozygote carriers [56]. Nonetheless, recent population studies revealed that some ATM polymorphisms have a protective role, while other studies reported a damaging effect [59,60,61,62]. There are even controversial observations for the same polymorphism [26,27,59,60,63]. Despite these controversies, consistent ATM variants (ATM p.P1054R-rs1800057- and rs149711770) were recently described in families with FNMTC and other cancers (as kidney, lung, stomach, and prostate) [11]. Nonetheless, in A-T Syndrome´s patient, the only screening recommended is for breast cancer [15].
2.6. DICER 1 Syndrome and miRNA Processing
A non-toxic multinodular goiter (MNG) is frequently diagnosed in the adult population and studies correlate the presence of MNG and the development of differentiated thyroid cancer [14,64,65]. On the other hand, familial cases of MNG are a common characteristic associated with the DICER1 syndrome (OMIM #601200), which predisposes patients to thyroid cancer [66], and other types of tumors such as Sertoli-Leydig cell tumors of the ovary (SLCT) [20] and pleuropulmonary blastomas [21].
DICER is an endonuclease essential for the maturation of microRNAs (miRNAs), small non-coding RNAs with ~22 nt, that block mRNA translation post-transcriptionally by binding to the 3′-UTR (untranslated region) of target mRNAs, and tightly controlling cell signaling and cell biology [67]. A mutation in dicer1, ribonuclease III (DICER1) gene, especially those present in the ribonuclease domain, leads to DICER loss of function and downregulation of microRNA levels [20,68]. The correct control of miRNA expression is essential for the development of a functional thyroid gland [69]. Studies with transgenic mice with dysfunctional DICER lead to disturbance of thyroid architecture, cell proliferation and disarrangement of follicular structures, and loss of differentiation [70,71], indicating the influence of DICER loss in thyroid tumorigenesis.
A familial approach to investigate the risk of thyroid malignancy in DICER1 syndrome patients revealed a 16-fold higher risk of development of thyroid cancer when DICER1 is mutated compared to non-mutated patients [66]. Thus, there is a suggestion to monitor the thyroid status by a thyroid ultrasound every two-three years in patients after the age of eight [29,72]. Enforced evidence of DICER1 mutation with familial thyroid cancer was also shown in a study with six individuals of the same family harboring DICER1 mutation (c.5441C>T, p.S1814L) and multiple cases of differentiated thyroid cancer and MNG [73].
The Cancer Genome Atlas (TCGA) database shows DICER1 mutation in 0.8% of patients with PTC/PDTC (p.E1813G, p.D1810H, p.E1813K, p.R1906S, p.M1402T) [74,75,76,77,78]. A recent study revealed high prevalence of DICER1 mutations in pediatric-adolescent poorly differentiated thyroid cancer (83%) at a hotspot in the metal-ion binding sites of the RNase IIIb domain of DICER1 (c.5113G>A, p.E1705K, c.5125G>A, p.D1709N (rs1595331264), c.5137G>A, p.1713Y, c.5437G>A, p.E1813K, c.5437G>C, p.E1813Q) [79]. Another study linked hotspot DICER1 mutations to pediatric PTC (c.5125G>A p.D1709N, c.5428G>T p.D1801Y, c.5438A>G p.E1813G, c.5439G>C p.E1813D) with increased incidence in the patients that do not harbor MAPK classic alterations [80], suggesting a role for DICER1 mutation detection in thyroid tumors. A recent study detected DICER1 (c.5429A>T, p.D1810V, c.5437G>A, p.E1813K) and drosha ribonuclease III (DROSHA) mutation (c.2943C>T, p.S981S, c.3597C>T, p.Y1199Y (rs61748189)) in benign follicular adenoma, even though DICER1 mutations were not detected in a follicular variant of PTC that harbored HRAS mutations [68]. On the other hand, a recent study associated MAPK alterations with germline mutations in DICER1 [81]. Altogether, these studies suggest that DICER1 haploinsufficiency is associated with thyroid tumorigenesis.
DROSHA is another endonuclease of miRNA processing machinery and acts together with DGCR8 to form the Microprocessor complex to excise the precursor miRNA out of the primary transcript in the nucleus [67]. Then, DICER acts in the next step in the cytoplasm and cleaves the precursor miRNA to form mature functional miRNAs. In a similar extent to DICER1 mutations, DGCR8 mutations were also detected in familial cases of MNG and are associated with schwannoma [82]. Altogether, these studies indicate the essential role of proper miRNA processing and expression for thyroid gland physiology.
2.7. Li-Fraumeni Syndrome
The Li-Fraumeni syndrome is caused by a heterozygous mutation in TP53 and is typically characterized by soft tissue and bone sarcomas, breast cancers, central nervous system tumors, leukemia, and adrenal tumors. p53 interacts with a complex network and drives DNA repair, cell-cycle arrest, senescence, or apoptosis when it is phosphorylated by DNA damage response (DDR) kinases [13,83]. The PTC occurs in 10% of Li-Fraumeni syndrome patients, mainly when associated with TP53 mutation p.R337H [24]. Therefore, imaging screening for thyroid malignancy in Li-Fraumeni families has been advocated [24].
3. Non-Syndromic FNMTC
Even if FNMTC comprises only 3–9% of all thyroid cancer, the first-degree relatives of NMTC have an 8-12-fold increased risk of developing the disease [84,85]. Non-syndromic FNMTC comprises 95% of all FNMTC and is defined by two or more first-degree relatives present with NMTC without associated syndromes. Moreover, the transmission pattern is not yet well defined, which seems to be autosomal dominant in most cases. Like sporadic NMTC, more than 85% are PTC, approximately 10% are FTC, and around 5% are anaplastic thyroid cancer. Furthermore, FNMTC is more aggressive, presents with nodal disease, and recurs more often. In addition, thyroid cancer tends to occur earlier in subsequent generations in FNMTC, called the anticipation phenomenon [2,86,87].
3.1. Linkage Analysis
From 1997 to 2006, the linkage analysis was the main method to study the familial condition. Using this approach, a positive logarithm of odds (LOD) would mean a high likelihood that locus cosegregates with the FNMTC trait, which is a linkage. In this way, several loci were associated with non-syndromic FNMTC (Table 2).
3.1.1. TCO Locus (19p13.2)
The ‘thyroid carcinoma, nonmedullary, with cell oxyphilia’ (TCO) locus was identified in a French family with oxyphilic thyroid cancer in the short arm of chromosome 19 (19p13.2). This region includes several genes, such as ICAM1 gene, which is overexpressed in thyroid cancer cells, and the JunB proto-oncogene, AP-1 transcription factor subunit (JUNB) [6]. However, some other genes in the locus, such as several zinc-finger-protein genes, were not yet identified. Moreover, the TCO locus does not seem to be involved in the majority of oxyphilic sporadic NMTC. An additional Tyrolean family with high LOD in the same locus was also described [93].
3.1.2. PRN1 Locus (1q21)
Papillary thyroid cancer is associated with papillary renal cancer. Linkage analysis identified this locus with the highest LOD of 3.58 in a family with three generations affected by PTC and papillary renal carcinoma. MET proto-oncogene, receptor tyrosine kinase (MET) mutations, frequently associated with familial papillary renal cancer, and mutations associated with other thyroid cancer syndromes were excluded [8]. However, this finding was limited to this family.
3.1.3. NMTC1 Locus (2q21)
This locus was described in a large Tasmanian family study [9], and when the authors further analyzed 17 families with FNMTC, they found an LOD heterogeneity of 4.17. At that time, it was hypothesized that multiple environmental and genetic causes could be involved in the pathogenesis of FNMTC [93].
3.1.4. q32 Locus (an Enhancer of Unknown Function)
A rare mutation in 4q32 was found in the linkage analysis and targeted deep sequencing in a large family with four individuals with benign thyroid disease, nine PTC patients, and one anaplastic thyroid cancer (ATC) patient. This nucleotide exchange in chr4:165491559 (GRCh37/hg19), named 4q32A>C, is in a highly conserved region. The chromatin immunoprecipitation (ChIP) assays showed that both POU2F1 and YY1 transcription factors related to specific thyroid genes and thyroid development bind to this region. As consequence of the allele’s change, a decrease of both POUF2 and YY1 bindings were observed. Transcription factors’ disruption has already been associated with cancer [91].
3.1.5. 6q22 Locus
The finding of 6q22 locus with LOD + 3.30 was observed in 38 families of FNMTC by linkage analysis and a genome-wide SNP array [89]. However, no further studies have confirmed this locus in additional families.
3.1.6. 8p23.1–p22 Locus
A locus associated with FNMTC in a huge Portuguese family was identified by linkage analysis, with a maximum parametric haplotype-based LOD score of 4.41. Among 17 candidate genes in the locus (PPP1R3B, MIRN597, MIRN124A1, MSRA, C8orf74, SOX7, PINX1, MIRN598, C8orf15, C8orf16, MTMR9, C8orf13, NEIL2, CTSB, DUB3, DLC1, TUSC3), no deleterious alteration was detected in the genes’ coding region. [10].
3.1.7. 8q24 Locus, a lncRNA inside the Thyroglobulin (TG) Gene
Linkage analysis was also performed in a group of 26 families of PTC [90], which revealed a LOD of + 1.3 in a locus that harbors TG and SLA (Src like adaptor) genes. However, no polymorphism or mutation was found in the coding genes, suggesting that this alteration could be associated with a lncRNA related to the TG gene.
3.1.8. SRGAP1 (12q14 Locus)
The study of 38 families with FNMTC by genome-wide linkage analysis indicated a high peak in 12q14 in 55% (21 of 38), but with a modest OR = 1.21 (p = 0.0008). Nonetheless, it was observed six different germline mutations/variants in the SRGAP1 gene (c.447A>C, p.Q149H, c.823G>A, p.A275T, c.1534G>A, p.V512I, rs74691643, c.1849C>T, p.R617C, rs114817817, c.2274T>C, p.S758S, rs789722, c.2624A>G, p.H875R, rs61754221). In vitro functional testing in thyroid cancer cells showed decreased GTPase activating protein (GAP) activity in two of these SGARP1 polymorphisms (Q149H and R617C). The SRGAP1 could mediate tumorigenesis by interacting with CDC42 [88], which is a common signal transduction convergence point of many signaling pathways and can play a role in thyroid cancer cell migration via RAGE/Dia-1 signaling [94].
3.1.9. NKX2-1 (14q13.3 Locus)
3.1.10. MNG1 Locus (14q32)-DICER1
The MNG1 (OMIM # 138800) locus was revealed by linkage analysis in families with multinodular goiter and NMTC [7]. Furthermore, it was observed that MNG1 corresponded to DICER1 gene, related to microRNA biogenesis (described in “Syndromic causes of non-medullary thyroid cancer” section).
3.2. Genome-Wide Linkage Analysis in the Population of PTC Patients
The sequencing of the genome by NGS) uncovered the genetic variation and the potential association with several pathologies, including cancer. In particular, the GWAS (genome-wide association study) revealed numerous SNPs in the genes related to thyroid physiology and tumorigenesis (Table 3) [12].
3.2.1. FOXE1/PTCSC2
Located in 9q22.3 and close to the forkhead box E1 (FOXE1) gene, rs965513 conferred an increased risk for thyroid cancer and was named ‘papillary thyroid carcinoma susceptibility candidate 2’ (PTCSC2) gene. The carriers of rs965513 (homozygous of A allele present a 3.1-fold increased risk for thyroid cancer in large European series [98]. The same polymorphism rs965513 was observed in Japanese and Belarusian populations, but with an OR of 1.6-1.9 [104]. Similarly, a variant in the promoter region of the FOXE1 gene (rs1867277) was identified as a risk factor for PTC (OR = 1.49) in a Spanish series and further confirmed in an Italian one [105]. Subsequently, new studies showed a tumor suppressor effect of FOXE1 and demonstrated that rs1867277 is involved in differential recruitment of USF1/ USF2 transcription factors, which interferes with FOXE1 expression [12,106]. Moreover, myosin heavy chain-9 (MYH9) can bind and suppress the shared promoter of PTCSC2 and FOXE1 genes bilaterally (that includes rs1867277 region), an effect that is abolished by PTCSC2 that sequesters MYH9 [107]. Therefore, MYH9, which is a lncRNA binding protein, can also play a role in PTC susceptibility.
3.2.2. NKX2-1
A consistent finding in the 14q13.3 locus was rs944289. Located close to the NKX2-1 gene, this variant of uncertain significance (VUS) is in PTCSC3’s promoter region and regulates the lncRNA PTCSC3 expression by affecting the binding site of C/EBPα and C/EBPβ (PTCSC3 activators) [98,99,109,110]. PTCSC3 downregulates S100A4, reducing cell motility and invasiveness. Thus, PTCSC3 mutations could predispose to PTC through the S100A4 pathway [111]. Moreover, NKX2-1 mutation (c.1016C>T, p.A339V) was observed in a family with multinodular goiter and papillary thyroid cancer [87], but this was not confirmed by another FNMTC study [112].
3.2.3. NRG1
NRG1 polymorphisms produced an association signal in GWAS for thyroid cancer. NRG1 is highly expressed in the thyroid and participates in cell growth pathways, mainly via erb-b2 receptor tyrosin kinase (ERBB)/MAPK [113]. However, NRG1 expression is detected in follicular adenomas, suggesting they are linked to thyroid tumorigenesis [12].
3.2.4. DIRC3
Polymorphisms in the DIRC3 (disrupted in renal carcinoma 3) gene have also been found in thyroid cancer GWAS [12,102,103]. DIRC3 codifies a lncRNA that was first associated with renal cancer, suggesting a tumor suppressor role [101]. DIRC3 and IGFBP5 (insulin-like growth factor binding protein 5) tumor suppressors are within the same topologically associated domain. Moreover, it was observed that DIRC3 depletion induces an increased SOX10 (SRY-box transcription factor 10) repression of IGFBP5 in melanoma cell cultures, corroborating the tumor suppressor role of DIRC3 [114].
In addition, the TT variant of rs966423 (DIRC3, g.217445617C>T) has been associated with worse PTC presentation and prognosis. An increased tumor size, staging, lymph node involvement, and overall mortality was observed in the TT-haplotype [115]. In a Chinese series, rs966423 was also correlated to tumor invasion and multifocality [1]. Nevertheless, no difference in these parameters was observed in a Polish series [116].
3.2.5. Polygenic Contribution
Recently, an increased risk for PTC was associated with a cumulative number of deleterious polymorphisms detected in the same patient. Ten different polymorphisms (rs12129938, rs11693806, rs6793295, rs73227498, rs2466076, rs1588635, rs7902587, rs368187, rs116909374, and rs2289261) related to the PTC development were analyzed, and the presence of each of these SNPs increased the risk to PTC. Nevertheless, if a patient harbors all 10 variants at the same time, the risk of developing thyroid cancer is 6.9-fold greater than those with no variants [117].
3.2.6. Telomere Abnormalities
A decade ago, three independent groups observed that relative telomere length (RTL) is shorter in patients with FNMTC [118,119,120]. As telomerase controls the telomere length, one of these groups investigated TERC and hTERT (which form telomerase) alterations and observed the amplification of hTERT in patients’ leukocytes [118]. However, this finding was not confirmed subsequently [119,120]. In recent years, many alterations in the shelterin complex’s genes have been reported. The shelterin complex is formed by six proteins (POT1, ACD, TINF2, TERF1, TERF2, and TERF2IP), and protects the telomere from DDR mechanisms. Along with telomerase, this complex is vital for genomic stability because telomeric ends resemble DNA double breaks. Telomeric repeat binding factor 1 (TERF1, also known as TRF1), telomeric repeat binding factor 2 (TERF2, also known TRF2), and protection of telomeres 1 (POT1) directly recognize TTAGGG repeats. In contrast, adrenocortical dysplasia protein homolog (ACD, also known as TPP1), TERF1-interacting nuclear factor 2 (TINF2, also known as TIN2), and telomeric repeat binding factor 2 interacting protein (TERF2IP, also known as RAP1) form a complex that differentiates telomeres from sites of DNA damage.
TINF2 mutation was described in a family with melanoma and thyroid cancer predisposition. Functional analysis showed that mutated TINF2 was unable to activate TERF2, resulting in longer telomere lengths. All shelterin complex’s genes were screened in a subsequent 24 families with FNMTC, and two missense variants in TINF2 and ACD genes were found, but only the ACD variant was predicted as deleterious [121].
Another group reported a new mutation in POT1 (c.85G>T; p.V29L) [122] in an Italian FNMTC. POT1 disruptions can interfere with the interaction of the POT1-ACD complex. In agreement with these findings, another POT1 mutation (c.268A>G, p.K90E) was described in a family with a predisposition to several tumors (melanoma, breast, kidney, and thyroid cancer, pituitary tumor, and Cushing syndrome) [123]. Moreover, an association between the increased risk of thyroid cancer and the presence of an intronic variant of POT1 (rs58722976) was also observed in a cohort of childhood cancer survivors [124].
Altogether, it suggests that telomere abnormalities and shelterin complex genes alteration may influence the predisposition to the FNMTC.
3.2.7. miRNA
The miRNA-related SNPs affect the microRNA biogenesis and function. A large study evaluated approximately 80 families displaying Mendelian-like inheritance and found two candidate miRNA (let-7e and miR-181b). The variants of let-7e and miR-182b-2 were located at the 5′ end of 3p mature miRNA and the 3′ end of 5p mature miRNA, respectively, which downregulate the expression by impairing the miRNA processing [125]. The gain or loss of specific miRNAs is an important oncogenic event [69].
3.3. Whole Exome/Genome Sequence
The whole-exome sequence (WES) or the whole genome sequence (WGS) of family members with FNMTC is another strategy besides the GWAS in large populations of differentiated thyroid cancer (DTC). Using this approach, an enormous number of variants is detected, demanding some criteria to filter and select the candidate variants. In general, minor allele frequency (MAF), the expression in thyroid and predictor functions (i.e., SIFT, PolyPhen, CADD, and others) are used as filters. Variants related to cancer pathways can also be used as filters. Since the application of this strategy has been consolidated for genetic studies in recent years, some authors have proposed new variants involved in FNMTC. Many are still under validation.
3.3.1. SRRM2
3.3.2. NOP53
The presence of rs78530808 (NOP53, c.91G>C, p.D31H) was observed in one family with FNMTC when using a less strict filter than other studies (MAF < 2%) [116]. NOP53 participates in ribosome biogenesis and regulates the p53 activation in the case of ribosome biogenesis perturbation. The variant c.91G>C was also identified in three out of 44 families with FNMTC [127]. In the tumor samples, NOP53 expression was increased when compared to the adjacent normal tissue. Furthermore, NOP53 knockdown inhibited cell proliferation and colony formation in vitro [127]. Altogether, these findings suggested that this variant could have an oncogenic role in thyroid tumorigenesis [127].
3.3.3. HABP2
HABP2 variant is an excellent example to describe how careful we should be with possible false-positive findings. The variant G534E was described in a family with seven members with PTC [128]. However, this finding was severely criticized later by other researchers. Even though it seemed the right candidate in the beginning, further studies did not confirm it in other populations. Furthermore, since its MAF is high in the European population, we would expect a higher incidence of FNMTC [129]. Besides, the prevalence of this same variant was similar among patients with FNMTC, sporadic PTC, and controls [130,131].
3.4. Candidate Variants Associated with FNMTC
Recently, different groups have pinpointed a list of candidate variants in FNMTC. A Korean study identified seven candidate variants localized in ANO7, CAV2, KANK1, PIK3CB, PKD1L1, PTPRF, and RHBDD2 genes in a family with four patients with PTC [132]. In addition, a Brazilian group reported seven new variants located in FKBP10, PLEKHG5, P2RX5, SAPCD1, ANXA3, NTN4, and SERPINA1 [133].
In a large series including 17 families with isolated FNMTC and FNMTC associated with other malignancies, 41 rare candidate variants were identified in TDRD6, IDE, TINF2, RNF213, AGK, NHLH1, TMCC1, ALB, THBS4, C5orf15, KLH3, FGFR4, SMARCD3, GPR107, NSMF, SVIL, EIF3, RNF169, NFRB, CIS, CDH11, EDC4, FOXA3, CDS2, NAPB, SALL4, ATG14, UNC79, LZTR1, ATP13A2, CTDSP1, MAPKAPK3, AARS, KDSR, ZNF302, ZNF17, ITGAD, FGD6, PDPR, and EFCAB8 genes. Cancer susceptibility genes (CHEK2, PRF1, ATM, AKAP13, SLC26A11) were also observed [11]. As described before, the authors further correlated the presence of TINF2 (a shelterin gene) to families with PTC and melanoma.
4. Conclusions
It was expected that the advent of new technologies of genome study would shed new light on the genetic predisposition of FNMTC. The NGS certainly did shed light on a whole new spectrum of variants and pointed to the co-occurrence of several variants in FNMTC. However, the limiting point in this scenario is the lack of a detailed in vitro validation that could precisely identify the contribution of each variant for the complex FNMTC entity. Moreover, the expansion of already known genetic data in multiple cohorts is essential to establish their role in FNMTC carcinogenesis.
Author Contributions
E.T.K. conceived the idea; F.Y.M., C.S.F., G.A.d.C. and E.T.K. were involved in planning, writing, and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financed in part by the CNPq (National Council for Scientific and Technological Development-Brazil) grant 430756/2018-6 (E.T.K.).
Acknowledgments
We gratefully acknowledge the CNPq by the grant 308331/2017-6 (E.T.K.), FAPESP (The São Paulo Research Foundation-Brazil) by the grant 2020/10403-9 (C.S.F.), and the scholarships from CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brazil): PDSE 88881.362254/2019-01 (F.Y.M.) and PNPD 88887.374682/2019-00 (C.S.F.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AD | autosomal dominant |
| AR | autosomal recessive |
| ATC | anaplastic thyroid cancer |
| CMVPTC | cribriform-morular variant of PTC |
| cPTC | classical PTC |
| DDR | DNA damage response |
| DTC | differentiated thyroid cancer |
| FA | follicular adenoma |
| FAP | familial adenomatous polyposis |
| FNMTC | familial nonmedullary thyroid cancer |
| FTC | follicular thyroid cancer |
| FVPTC | follicular variant of PTC |
| GWAS | genome-wide association studies |
| HR | hazard ratio |
| LOD | logarithm of odds |
| MAF | minor allele frequency |
| miRNAs | microRNAs |
| MNG | multinodular goiter |
| MTC | medullary thyroid cancer |
| NGS | next generation sequencing |
| NMTC | nonmedullary thyroid cancer |
| PTC | papillary thyroid cancer |
| SNPs | single nucleotide polymorphisms |
| TCGA | The Cancer Genome Atlas |
| VUS | variant of uncertain significance |
| WES | whole exome sequence |
| WGS | whole genome sequence |
References
- Hincza, K.; Kowalik, A.; Kowalska, A. Current Knowledge of Germline Genetic Risk Factors for the Development of Non-Medullary Thyroid Cancer. Genes 2019, 10, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilmette, J.; Nosé, V. Hereditary and familial thyroid tumours. Histopathology 2017, 72, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firminger, H.I.; Skelton, F.R. Carcinoma of the thyroid: Papillary adenocarcinoma occurring in twins and a case of Hürthle cell carcinoma; tumor conference. J. Kans. Med. Soc. 1953, 54, 427–432. [Google Scholar] [PubMed]
- Sippel, R.S.; Caron, N.R.; Clark, O.H. An evidence-based approach to familial nonmedullary thyroid cancer: Screening, clinical management, and follow-up. World J. Surg. 2007, 31, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Mazeh, H.; Sippel, R.S. Familial nonmedullary thyroid carcinoma. Thyroid 2013, 23, 1049–1056. [Google Scholar] [CrossRef]
- Canzian, F.; Amati, P.; Harach, H.R.; Kraimps, J.-L.; Lesueur, F.; Barbier, J.; Levillain, P.; Romeo, G.; Bonneau, D. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am. J. Hum. Genet. 1998, 63, 1743–1748. [Google Scholar] [CrossRef] [Green Version]
- Bignell, G.R.; Canzian, F.; Shayeghi, M.; Stark, M.; Shugart, Y.Y.; Biggs, P.; Mangion, J.; Hamoudi, R.; Rosenblatt, J.; Buu, P.; et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am. J. Hum. Genet. 1997, 61, 1123–1130. [Google Scholar] [CrossRef] [Green Version]
- Malchoff, C.D.; Sarfarazi, M.; Tendler, B.; Forouhar, F.; Whalen, G.; Joshi, V.; Arnold, A.; Malchoff, D.M. Papillary thyroid carcinoma associated with papillary renal neoplasia: Genetic linkage analysis of a distinct heritable tumor syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 1758–1764. [Google Scholar] [CrossRef]
- McKay, J.D.; Lesueur, F.; Jonard, L.; Pastore, A.; Williamson, J.; Hoffman, L.; Burgess, J.; Duffield, A.; Papotti, M.; Stark, M.; et al. Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am. J. Hum. Genet. 2001, 69, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Cavaco, B.M.; Batista, P.F.; Sobrinho, L.G.; Leite, V. Mapping a new familial thyroid epithelial neoplasia susceptibility locus to chromosome 8p23.1-p22 by high-density single-nucleotide polymorphism genome-wide linkage analysis. J. Clin. Endocrinol. Metab. 2008, 93, 4426–4430. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liyanarachchi, S.; Miller, K.E.; Nieminen, T.T.; Comiskey, D.F., Jr.; Li, W.; Brock, P.; Symer, D.E.; Akagi, K.; DeLap, K.E.; et al. Identification of Rare Variants Predisposing to Thyroid Cancer. Thyroid 2019, 29, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Saenko, V.A.; Rogounovitch, T.I. Genetic Polymorphism Predisposing to Differentiated Thyroid Cancer: A Review of Major Findings of the Genome-Wide Association Studies. Endocrinol. Metab. 2018, 33, 164–174. [Google Scholar] [CrossRef]
- Carbone, M.; Arron, S.T.; Beutler, B.; Bononi, A.; Cavenee, W.; Cleaver, J.E.; Croce, C.M.; D’Andrea, A.; Foulkes, W.D.; Gaudino, G.; et al. Tumour predisposition and cancer syndromes as models to study gene-environment interactions. Nat. Rev. Cancer 2020, 20, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [Green Version]
- Van Os, N.J.; Roeleveld, N.; Weemaes, C.M.R.; Jongmans, M.C.J.; Janssens, G.O.; Taylor, A.M.R.; Hoogerbrugge, N.; Willemsen, M.A.A.P. Health risks for ataxia-telangiectasia mutated heterozygotes: A systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy-Perez, B.; Janin, N.; Ossian, K.; Laugé, A.; Croquette, M.F.; Griscelli, C.; Debré, M.; Bressac-de-Paillerets, B.; Aurias, A.; Stoppa-Lyonnet, D.; et al. Cancer risk in heterozygotes for ataxia-telangiectasia. Int. J. Cancer 2001, 93, 288–293. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Faucz, F.R.; Voutetakis, A.; Stratakis, C.A. Carney Complex. Exp. Clin. Endocrinol. Diabetes 2019, 127, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Gammon, A.; Jasperson, K.; Champine, M. Genetic basis of Cowden syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2016, 9, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Frio, T.R.; Bahubeshi, A.; Kanellopoulou, C.; Hamel, N.; Niedziela, M.; Sabbaghian, N.; Pouchet, C.; Gilbert, L.; O’Brien, P.K.; Serfas, K.; et al. DICER1 Mutations in Familial Multinodular Goiter With and Without Ovarian Sertoli-Leydig Cell Tumors. JAMA 2011, 305, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 Mutations in Familial Pleuropulmonary Blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomoda, C.; Miyauchi, A.; Uruno, T.; Takamura, Y.; Ito, Y.; Miya, A.; Kobayashi, K.; Matsuzuka, F.; Kuma, S.; Kuma, K.; et al. Cribriform-morular variant of papillary thyroid carcinoma: Clue to early detection of familial adenomatous polyposis-associated colon cancer. World J. Surg. 2004, 28, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, T.T.; Walker, C.J.; Olkinuora, A.; Genutis, L.K.; O’Malley, M.; Wakely, P.E.; LaGuardia, L.; Koskenvuo, L.; Arola, J.; Lepistö, A.H.; et al. Thyroid Carcinomas That Occur in Familial Adenomatous Polyposis Patients Recurrently Harbor Somatic Variants in APC, BRAF, and KTM2D. Thyroid 2020, 30, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Formiga, M.N.D.C.; De Andrade, K.C.; Kowalski, L.P.; Achatz, M.I. Frequency of Thyroid Carcinoma in Brazilian TP53 p.R337H Carriers with Li Fraumeni Syndrome. JAMA Oncol. 2017, 3, 1400–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereda, C.M.; Lesueur, F.; Pertesi, M.; Robinot, N.; Lence-Anta, J.J.; Turcios, S.; Velasco, M.; Chappe, M.; Infante, I.; Bustillo, M.; et al. Common variants at the 9q22.33, 14q13.3 and ATM loci, and risk of differentiated thyroid cancer in the Cuban population. BMC Genet. 2015, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Maillard, S.; Damiola, F.; Clero, E.; Pertesi, M.; Robinot, N.; Rachédi, F.; Boissin, J.-L.; Sebbag, J.; Shan, L.; Bost-Bezeaud, F.; et al. Common variants at 9q22.33, 14q13.3, and ATM loci, and risk of differentiated thyroid cancer in the French Polynesian population. PLoS ONE 2015, 10, e0123700. [Google Scholar] [CrossRef]
- Tan, M.-H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.-L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011, 88, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.A.P.; Rednam, S.P.; Kamihara, J.; Doros, L.; Achatz, M.I.; Wasserman, J.D.; Diller, L.R.; Brugières, L.; Druker, H.; Schneider, K.A.; et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, e76–e82. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Kiyotani, K.; Yew, P.Y.; Kato, T.; Tamura, K.; Yap, K.L.; Nielsen, S.M.; Mester, J.L.; Eng, C.; Nakamura, Y.; et al. Germline PARP4 mutations in patients with primary thyroid and breast cancers. Endocr. Relat. Cancer 2016, 23, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Yehia, L.; Ni, Y.; Sesock, K.; Niazi, F.; Fletcher, B.; Chen, H.J.L.; LaFramboise, T.; Eng, C. Unexpected cancer-predisposition gene variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome patients without underlying germline PTEN mutations. PLoS Genet. 2018, 14, e1007352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngeow, J.; Ni, Y.; Tohme, R.; Chen, F.S.; Bebek, G.; Eng, C. Germline alterations in RASAL1 in Cowden syndrome patients presenting with follicular thyroid cancer and in individuals with apparently sporadic epithelial thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1316–E1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Costa, H.A.; Leitner, M.G.; Sos, M.L.; Mavrantoni, A.; Rychkova, A.; Johnson, J.R.; Newton, B.W.; Yee, M.C.; De La Vega, F.M.; Ford, J.M.; et al. Discovery and functional characterization of a neomorphic PTEN mutation. Proc. Natl. Acad. Sci. USA 2015, 112, 13976–13981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Ringel, M.D.; Hayre, N.; Saito, J.; Saunier, B.; Schuppert, F.; Burch, H.; Bernet, V.; Burman, K.D.; Kohn, L.D.; Saji, M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001, 61, 6105–6111. [Google Scholar]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. 2013, 105, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Pepe, S.; Korbonits, M.; Iacovazzo, D. Germline and mosaic mutations causing pituitary tumours: Genetic and molecular aspects. J. Endocrinol. 2019, 240, R21–R45. [Google Scholar] [CrossRef] [Green Version]
- Griffin, K.J.; Kirschner, L.S.; Matyakhina, L.; Stergiopoulos, S.; Robinson-White, A.; Lenherr, S.; Weinberg, F.D.; Claflin, E.; Meoli, E.; Cho-Chung, Y.S.; et al. Down-regulation of regulatory subunit type 1A of protein kinase A leads to endocrine and other tumors. Cancer Res. 2004, 64, 8811–8815. [Google Scholar] [CrossRef] [Green Version]
- Sandrini, F.; Matyakhina, L.; Sarlis, N.J.; Kirschner, L.S.; Farmakidis, C.; Gimm, O.; Stratakis, C.A. Regulatory subunit type I-α of protein kinase A (PRKAR1A): A tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer 2002, 35, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Pringle, D.R.; Yin, Z.; Lee, A.A.; Manchanda, P.K.; Yu, L.; Parlow, A.F.; Jarjoura, D.; La Perle, K.M.D.; Kirschner, L.S. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocrine-Related Cancer 2012, 19, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kari, S.; Vasko, V.V.; Priya, S.; Kirschner, L.S. PKA Activates AMPK Through LKB1 Signaling in Follicular Thyroid Cancer. Front. Endocrinol. 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Biomarkers Prev. 1996, 5, 239–246. [Google Scholar] [PubMed]
- Muftuoglu, M.; Oshima, J.; von Kobbe, C.; Cheng, W.H.; Leistritz, D.F.; Bohr, V.A. The clinical characteristics of Werner syndrome: Molecular and biochemical diagnosis. Hum. Genet. 2008, 124, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Shamanna, R.A.; Lu, H.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Sugano, H.; Matsumoto, T.; Furuichi, Y.; Miller, R.W.; Goto, M. Unusual features of thyroid carcinomas in Japanese patients with Werner syndrome and possible genotype-phenotype relations to cell type and race. Cancer 1999, 85, 1345–1352. [Google Scholar] [CrossRef]
- Uchino, S.; Noguchi, S.; Yamashita, H.; Yamashita, H.; Watanabe, S.; Ogawa, T.; Tsuno, A.; Murakami, A.; Miyauchi, A. Mutational analysis of the APC gene in cribriform-morula variant of papillary thyroid carcinoma. World J. Surg. 2006, 30, 775–779. [Google Scholar] [CrossRef]
- De Herreros, A.G.; Duñach, M. Intracellular Signals Activated by Canonical Wnt Ligands Independent of GSK3 Inhibition and β-Catenin Stabilization. Cells 2019, 8, 1148. [Google Scholar] [CrossRef] [Green Version]
- Heinen, C.D. Genotype to phenotype: Analyzing the effects of inherited mutations in colorectal cancer families. Mutat. Res. 2010, 693, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Giannelli, S.M.; McPhaul, L.; Nakamoto, J.; Gianoukakis, A.G. Familial adenomatous polyposis-associated, cribriform morular variant of papillary thyroid carcinoma harboring a K-RAS mutation: Case presentation and review of molecular mechanisms. Thyroid 2014, 24, 1184–1189. [Google Scholar] [CrossRef]
- Iwama, T.; Konishi, M.; Iijima, T.; Yoshinaga, K.; Tominaga, T.; Koike, M.; Miyaki, M. Somatic mutation of the APC gene in thyroid carcinoma associated with familial adenomatous polyposis. Jpn. J. Cancer Res. 1999, 90, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, M.; Iijima, T.; Ishii, R.; Hishima, T.; Mori, T.; Yoshinaga, K.; Takami, H.; Kuroki, T.; Iwama, T. Molecular Evidence for Multicentric Development of Thyroid Carcinomas in Patients with Familial Adenomatous Polyposis. Am. J. Pathol. 2000, 157, 1825–1827. [Google Scholar] [CrossRef] [Green Version]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achatz, M.I.; Porter, C.C.; Brugières, L.; Druker, H.; Frebourg, T.; Foulkes, W.D.; Kratz, C.P.; Kuiper, R.P.; Hansford, J.R.; Hernandez, H.S.; et al. Cancer Screening Recommendations and Clinical Management of Inherited Gastrointestinal Cancer Syndromes in Childhood. Clin. Cancer Res. 2017, 23, e107–e114. [Google Scholar] [CrossRef] [Green Version]
- Dombernowsky, S.L.; Weischer, M.; Allin, K.H.; Bojesen, S.E.; Tybjirg-Hansen, A.; Nordestgaard, B.G. Risk of cancer by ATM missense mutations in the general population. J. Clin. Oncol. 2008, 26, 3057–3062. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM: Expanding roles as a chief guardian of genome stability. Exp. Cell Res. 2014, 329, 154–161. [Google Scholar] [CrossRef]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37–38, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Akulevich, N.M.; Saenko, V.A.; Rogounovitch, T.I.; Drozd, V.M.; Lushnikov, E.F.; Ivanov, V.K.; Mitsutake, N.; Kominami, R.; Yamashita, S. Polymorphisms of DNA damage response genes in radiation-related and sporadic papillary thyroid carcinoma. Endocr. Relat. Cancer 2009, 16, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Morari, E.C.; Wei, Q.; Sturgis, E.M.; Ward, L.S. Functional variations in the ATM gene and susceptibility to differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Shi, J.; Qiu, S.; Qiao, Y.; Zhang, X.; Cheng, Y.; Liu, Y. Association between ATM rs1801516 polymorphism and cancer susceptibility: A meta-analysis involving 12,879 cases and 18,054 controls. BMC Cancer 2018, 18, 1060. [Google Scholar] [CrossRef] [Green Version]
- Song, C.M.; Kwon, T.K.; Park, B.L.; Ji, Y.B.; Tae, K. Single nucleotide polymorphisms of ataxia telangiectasia mutated and the risk of papillary thyroid carcinoma. Environ. Mol. Mutagen. 2015, 56, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Wójcicka, A.; Czetwertyńska, M.; Świerniak, M.; Długosińska, J.; Maciąg, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Płoski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitahara, C.M.; Farkas, D.K.R.; Jørgensen, J.O.L.; Cronin-Fenton, D.; Sørensen, H.T. Benign Thyroid Diseases and Risk of Thyroid Cancer: A Nationwide Cohort Study. J. Clin. Endocrinol. Metab. 2018, 103, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Chen, X.; Schneider, D.F.; Broome, J.T.; Sippel, R.S.; Chen, H.; Solórzano, C.C. Cancer after Thyroidectomy: A Multi-Institutional Experience with 1,523 Patients. J. Am. Coll. Surg. 2013, 216, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.E.; Bauer, A.J.; Schultz, K.A.P.; Doros, L.; DeCastro, R.M.; Ling, A.; Lodish, M.B.; Harney, L.A.; Kase, R.G.; Carr, A.G.; et al. Quantification of Thyroid Cancer and Multinodular Goiter Risk in the DICER1 Syndrome: A Family-Based Cohort Study. J. Clin. Endocrinol. Metab. 2017, 102, 1614–1622. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Poma, A.M.; Condello, V.; Denaro, M.; Torregrossa, L.; Elisei, R.; Vitti, P.; Basolo, F. DICER1 somatic mutations strongly impair miRNA processing even in benign thyroid lesions. Oncotarget 2019, 10, 1785–1797. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. MicroRNAs in thyroid development, function and tumorigenesis. Mol. Cell. Endocrinol. 2017, 456, 44–50. [Google Scholar] [CrossRef]
- Rodriguez, W.; Jin, L.; Janssens, V.; Pierreux, C.; Hick, A.-C.; Urizar, E.; Costagliola, S. Deletion of the RNaseIII Enzyme Dicer in Thyroid Follicular Cells Causes Hypothyroidism with Signs of Neoplastic Alterations. PLoS ONE 2012, 7, e29929. [Google Scholar] [CrossRef]
- Frezzetti, D.; Reale, C.; Calì, G.; Nitsch, L.; Fagman, H.; Nilsson, O.; Scarfò, M.; De Vita, G.; Di Lauro, R. The microRNA-Processing Enzyme Dicer Is Essential for Thyroid Function. PLoS ONE 2011, 6, e27648. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, M.M.; Jha, P.; Schultz, K.A.P.; Sheil, A.; Harris, A.K.; Bauer, A.J.; Field, A.L.; Geller, J.; Hill, D.A. DICER1Mutations and Differentiated Thyroid Carcinoma: Evidence of a Direct Association. J. Clin. Endocrinol. Metab. 2016, 101, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org (accessed on 16 August 2020).
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Chernock, R.D.; Rivera, B.; Borrelli, N.; Hill, D.A.; Fahiminiya, S.; Shah, T.; Chong, A.-S.; Aqil, B.; Mehrad, M.; Giordano, T.J.; et al. Poorly differentiated thyroid carcinoma of childhood and adolescence: A distinct entity characterized by DICER1 mutations. Mod. Pathol. 2020, 33, 1264–1274. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Sabbaghian, N.; Fahiminiya, S.; Chami, R.; Mete, O.; Acker, M.; Wu, M.K.; Shlien, A.; De Kock, L.; Foulkes, W.D. DICER1 Mutations Are Frequent in Adolescent-Onset Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 2009–2015. [Google Scholar] [CrossRef] [Green Version]
- Canberk, S.; Ferreira, J.C.; Pereira, L.; Batısta, R.; Vieira, A.F.; Soares, P.; Simões, M.S.; Máximo, V. Analyzing the Role of DICER1 Germline Variations in Papillary Thyroid Carcinoma. Eur. Thyroid J. 2020, 1–8. [Google Scholar] [CrossRef]
- Rivera, B.; Nadaf, J.; Fahiminiya, S.; Apellaniz-Ruiz, M.; Saskin, A.; Chong, A.-S.; Sharma, S.; Wagener, R.; Revil, T.; Condello, V.; et al. DGCR8 microprocessor defect characterizes familial multinodular goiter with schwannomatosis. J. Clin. Investig. 2020, 130, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldgar, D.E.; Easton, D.F.; Cannon-Albright, L.A.; Skolnick, M.H. Systematic Population-Based Assessment of Cancer Risk in First-Degree Relatives of Cancer Probands. J. Natl. Cancer Inst. 1994, 86, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Eng, C.; Chen, B. Familial Risks for Nonmedullary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2005, 90, 5747–5753. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.J. Clinical Behavior and Genetics of Nonsyndromic, Familial Nonmedullary Thyroid Cancer. Front. Horm. Res. 2013, 41, 141–148. [Google Scholar] [CrossRef]
- Capezzone, M.; Marchisotta, S.; Cantara, S.; Busonero, G.; Brilli, L.; Pazaitou-Panayiotou, K.; Carli, A.F.; Caruso, G.; Toti, P.; Capitani, S.; et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr. Relat. Cancer 2008, 15, 1075–1081. [Google Scholar] [CrossRef]
- He, H.; Bronisz, A.; Liyanarachchi, S.; Nagy, R.; Li, W.; Huang, Y.; Akagi, K.; Saji, M.; Kula, D.; Wojcicka, A.; et al. SRGAP1 Is a Candidate Gene for Papillary Thyroid Carcinoma Susceptibility. J. Clin. Endocrinol. Metab. 2013, 98, E973–E980. [Google Scholar] [CrossRef] [Green Version]
- Suh, I.; Filetti, S.; Vriens, M.R.; Guerrero, M.A.; Tumino, S.; Wong, M.; Shen, W.T.; Kebebew, E.; Duh, Q.-Y.; Clark, O.H. Distinct loci on chromosome 1q21 and 6q22 predispose to familial nonmedullary thyroid cancer: A SNP array-based linkage analysis of 38 families. Surgery 2009, 146, 1073–1080. [Google Scholar] [CrossRef]
- He, H.; Nagy, R.; Liyanarachchi, S.; Jiao, H.; Li, W.; Suster, S.; Kere, J.; De La Chapelle, A. A Susceptibility Locus for Papillary Thyroid Carcinoma on Chromosome 8q24. Cancer Res. 2009, 69, 625–631. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Li, W.; Wu, D.; Nagy, R.; Liyanarachchi, S.; Akagi, K.; Jendrzejewski, J.; Jiao, H.; Hoag, K.; Wen, B.; et al. Ultra-Rare Mutation in Long-Range Enhancer Predisposes to Thyroid Carcinoma with High Penetrance. PLoS ONE 2013, 8, e61920. [Google Scholar] [CrossRef]
- Ngan, E.S.W.; Lang, B.H.H.; Liu, T.; Shum, C.K.Y.; So, M.-T.; Lau, D.K.C.; Leon, T.Y.Y.; Cherny, S.S.; Tsai, S.Y.; Lo, C.-Y.; et al. A Germline Mutation (A339V) in Thyroid Transcription Factor-1 (TITF-1/NKX2.1) in Patients with Multinodular Goiter and Papillary Thyroid Carcinoma. J. Natl. Cancer Inst. 2009, 101, 162–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, J.D.; Thompson, D.B.; Lesueur, F.; Stankov, K.; Pastore, A.; Watfah, C.; Strolz, S.; Riccabona, G.; Moncayo, R.C.; Romeo, G.; et al. Evidence for interaction between the TCO and NMTC1 loci in familial non-medullary thyroid cancer. J. Med. Genet. 2004, 41, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE Mediates the Pro-Migratory Response of Extracellular S100A4 in Human Thyroid Cancer Cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.R.; Duffield, A.; Wilkinson, S.J.; Ware, R.; Greenaway, T.M.; Percival, J.; Hoffman, L. Two Families with an Autosomal Dominant Inheritance Pattern for Papillary Carcinoma of the Thyroid. J. Clin. Endocrinol. Metab. 1997, 82, 345–348. [Google Scholar] [CrossRef]
- Bakhsh, A.; Kirov, G.; Gregory, J.W.; Williams, E.D.; Ludgate, M. A new form of familial multi-nodular goitre with progression to differentiated thyroid cancer. Endocr.-Relat. Cancer 2006, 13, 475–483. [Google Scholar] [CrossRef]
- Franceschi, S.; Preston-Martin, S.; Maso, L.D.; Negri, E.; La Vecchia, C.; Mack, W.J.; McTiernan, A.; Kolonel, L.; Mark, S.D.; Mabuchi, K.; et al. A pooled analysis of case—Control studies of thyroid cancer. IV. Benign thyroid diseases. Cancer Causes Control. 1999, 10, 583–595. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Jonasson, J.G.; Sigurdsson, A.; Bergthorsson, J.T.; He, H.; Blondal, T.; Geller, F.; Jakobsdottir, M.; et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 2009, 41, 460–464. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Jonasson, J.G.; Masson, G.; He, H.; Jonasdottir, A.; Sigurdsson, A.; Stacey, S.N.; Johannsdottir, H.; et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat. Genet. 2012, 44, 319–322. [Google Scholar] [CrossRef]
- Takahashi, M.; Saenko, V.A.; Rogounovitch, T.I.; Kawaguchi, T.; Drozd, V.M.; Takigawa-Imamura, H.; Akulevich, N.M.; Ratanajaraya, C.; Mitsutake, N.; Takamura, N.; et al. The FOXE1 locus is a major genetic determinant for radiation-related thyroid carcinoma in Chernobyl. Hum. Mol. Genet. 2010, 19, 2516–2523. [Google Scholar] [CrossRef] [Green Version]
- Köhler, A.; Chen, B.; Gemignani, F.; Elisei, R.; Romei, C.; Figlioli, G.; Cipollini, M.; Cristaudo, A.; Bambi, F.; Hoffmann, P.; et al. Genome-Wide Association Study on Differentiated Thyroid Cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1674–E1681. [Google Scholar] [CrossRef] [Green Version]
- Son, H.-Y.; Hwangbo, Y.; Yoo, S.-K.; Im, S.-W.; Yang, S.D.; Kwak, S.-J.; Park, M.S.; Kwak, S.H.; Cho, S.W.; Ryu, J.S.; et al. Genome-wide association and expression quantitative trait loci studies identify multiple susceptibility loci for thyroid cancer. Nat. Commun. 2017, 8, 15966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudmundsson, J.; Thorleifsson, G.; Sigurdsson, J.K.; Stefansdottir, L.; Jonasson, J.G.; Gudjonsson, S.A.; Gudbjartsson, D.F.; Masson, G.; Johannsdottir, H.; Halldorsson, G.H.; et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 2017, 8, 14517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitski, A.V.; Rogounovitch, T.I.; Bychkov, A.; Takahashi, M.; Yoshiura, K.-I.; Mitsutake, N.; Kawaguchi, T.; Matsuse, M.; Drozd, V.M.; Demidchik, Y.; et al. Genotype Analyses in the Japanese and Belarusian Populations Reveal Independent Effects of rs965513 and rs1867277 but Do Not Support the Role of FOXE1 Polyalanine Tract Length in Conferring Risk for Papillary Thyroid Carcinoma. Thyroid 2017, 27, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, I.; Ruiz-Llorente, S.; Montero-Conde, C.; Inglada-Pérez, L.; Schiavi, F.; Leskelä, S.; Pita, G.; Milne, R.; Maravall, J.; Ramos, I.; et al. The Variant rs1867277 in FOXE1 Gene Confers Thyroid Cancer Susceptibility through the Recruitment of USF1/USF2 Transcription Factors. PLoS Genet. 2009, 5, e1000637. [Google Scholar] [CrossRef] [Green Version]
- Nikitski, A.; Saenko, V.; Shimamura, M.; Nakashima, M.; Matsuse, M.; Suzuki, K.; Rogounovitch, T.I.; Bogdanova, T.; Shibusawa, N.; Yamada, M.; et al. Targeted Foxe1 Overexpression in Mouse Thyroid Causes the Development of Multinodular Goiter But Does Not Promote Carcinogenesis. Endocrinology 2016, 157, 2182–2195. [Google Scholar] [CrossRef]
- Wang, Y.; He, H.; Li, W.; Phay, J.; Shen, R.; Yu, L.; Hancioglu, B.; De La Chapelle, A. MYH9 binds to lncRNA genePTCSC2and regulates FOXE1 in the 9q22 thyroid cancer risk locus. Proc. Natl. Acad. Sci. USA 2017, 114, 474–479. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.; Howarth, K.M.; Martin, L.; Gorman, M.; Mihai, R.; Moss, L.; Auton, A.; Lemon, C.; Mehanna, H.; Mohan, H.; et al. Thyroid cancer susceptibility polymorphisms: Confirmation of loci on chromosomes 9q22 and 14q13, validation of a recessive 8q24 locus and failure to replicate a locus on 5q24. J. Med. Genet. 2012, 49, 158–163. [Google Scholar] [CrossRef]
- Jendrzejewski, J.; He, H.; Radomska, H.S.; Li, W.; Tomsic, J.; Liyanarachchi, S.; Davuluri, R.V.; Nagy, R.; De La Chapelle, A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA 2012, 109, 8646–8651. [Google Scholar] [CrossRef] [Green Version]
- Goedert, L.; Plaça, J.R.; Fuziwara, C.S.; Machado, M.C.R.; Plaça, D.R.; Almeida, P.P.; Sanches, T.P.; Dos Santos, J.F.; Corveloni, A.C.; Pereira, I.E.G.; et al. Identification of Long Noncoding RNAs Deregulated in Papillary Thyroid Cancer and Correlated with BRAFV600E Mutation by Bioinformatics Integrative Analysis. Sci. Rep. 2017, 7, 1662. [Google Scholar] [CrossRef] [Green Version]
- Jendrzejewski, J.; Thomas, A.; Liyanarachchi, S.; Eiterman, A.; Tomsic, J.; He, H.; Radomska, H.S.; Li, W.; Nagy, R.; Sworczak, K.; et al. PTCSC3 Is Involved in Papillary Thyroid Carcinoma Development by Modulating S100A4 Gene Expression. J. Clin. Endocrinol. Metab. 2015, 100, E1370–E1377. [Google Scholar] [CrossRef] [Green Version]
- Cantara, S.; Capuano, S.; Formichi, C.; Pisu, M.; Capezzone, M.; Pacini, F. Lack of germline A339V mutation in thyroid transcription factor-1 (TITF-1/NKX2.1) gene in familial papillary thyroid cancer. Thyroid Res. 2010, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Li, W.; Liyanarachchi, S.; Wang, Y.; Yu, L.; Genutis, L.K.; Maharry, S.; Phay, J.E.; Shen, R.; Brock, P.; et al. The Role of NRG1 in the Predisposition to Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Coe, E.A.; Tan, J.Y.; Shapiro, M.; Louphrasitthiphol, P.; Bassett, A.R.; Marques, A.C.; Goding, C.R.; Vance, K.W. The MITF-SOX10 regulated long non-coding RNA DIRC3 is a melanoma tumour suppressor. PLoS Genet. 2019, 15, e1008501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Świerniak, M.; Wójcicka, A.; Czetwertyńska, M.; Długosińska, J.; Stachlewska, E.; Gierlikowski, W.; Kot, A.; Górnicka, B.; Koperski, Ł.; Bogdańska, M.; et al. Association between GWAS-Derived rs966423 Genetic Variant and Overall Mortality in Patients with Differentiated Thyroid Cancer. Clin. Cancer Res. 2016, 22, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Hińcza, K.; Kowalik, A.; Pałyga, I.; Walczyk, A.; Gąsior-Perczak, D.; Mikina, E.; Trybek, T.; Szymonek, M.; Gadawska-Juszczyk, K.; Zajkowska, K.; et al. Does the TT Variant of the rs966423 Polymorphism in DIRC3 Affect the Stage and Clinical Course of Papillary Thyroid Cancer? Cancers 2020, 12, 423. [Google Scholar] [CrossRef] [Green Version]
- Liyanarachchi, S.; Gudmundsson, J.; Ferkingstad, E.; He, H.; Jonasson, J.G.; Tragante, V.; Asselbergs, F.W.; Xu, L.; Kiemeney, L.A.; Netea-Maier, R.T.; et al. Assessing thyroid cancer risk using polygenic risk scores. Proc. Natl. Acad. Sci. USA 2020, 117, 5997–6002. [Google Scholar] [CrossRef]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Filetti, S.; De Santi, M.M.; Rossi, B.; Ronga, G.; Durante, C.; Pacini, F. Short Telomeres, Telomerase Reverse Transcriptase Gene Amplification, and Increased Telomerase Activity in the Blood of Familial Papillary Thyroid Cancer Patients. J. Clin. Endocrinol. Metab. 2008, 93, 3950–3957. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Bian, B.; Gesuwan, K.; Gulati, N.; Zhang, L.; Nilubol, N.; Kebebew, E. Telomere Length Is Shorter in Affected Members of Families with Familial Nonmedullary Thyroid Cancer. Thyroid 2013, 23, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Cantara, S.; Capuano, S.; Capezzone, M.; Benigni, M.; Pisu, M.; Marchisotta, S.; Pacini, F. Lack of Mutations of the Telomerase RNA Component in Familial Papillary Thyroid Cancer with Short Telomeres. Thyroid 2012, 22, 363–368. [Google Scholar] [CrossRef]
- He, H.; Li, W.; Comiskey, D.F.; Liyanarachchi, S.; Nieminen, T.T.; Wang, Y.; DeLap, K.E.; Brock, P.; De La Chapelle, A. A Truncating Germline Mutation of TINF2 in Individuals with Thyroid Cancer or Melanoma Results in Longer Telomeres. Thyroid 2020, 30, 204–213. [Google Scholar] [CrossRef]
- Srivastava, A.; Miao, B.; Skopelitou, D.; Kumar, V.; Kumar, A.; Paramasivam, N.; Bonora, E.; Hemminki, K.; Foersti, A.; Bandapalli, O.R. A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer. Cancers 2020, 12, 1441. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.L.-S.; Hattangady, N.; Lerario, A.M.; Williams, C.; Koeppe, E.; Quinonez, S.; Osborne, J.; Cha, K.B.; Else, T. A new POT1 germline mutation—expanding the spectrum of POT1-associated cancers. Fam. Cancer 2017, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.A.; Lupo, P.J.; Morton, L.M.; Yasui, Y.A.; Sapkota, Y.A.; Arnold, M.A.; Aubert, G.; Neglia, J.P.; Turcotte, L.M.; Leisenring, W.M.; et al. Genetic variation in POT1 and risk of thyroid subsequent malignant neoplasm: A report from the Childhood Cancer Survivor Study. PLoS ONE 2020, 15, e0228887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomsic, J.; Fultz, R.; Liyanarachchi, S.; Genutis, L.K.; Wang, Y.; Li, W.; Volinia, S.; Jazdzewski, K.; He, H.; Wakely, P.E., Jr.; et al. Variants in microRNA genes in familial papillary thyroid carcinoma. Oncotarget 2017, 8, 6475–6482. [Google Scholar] [CrossRef] [Green Version]
- Tomsic, J.; He, H.; Akagi, K.; Liyanarachchi, S.; Pan, Q.; Bertani, B.; Nagy, R.; Symer, D.E.; Blencowe, B.J.; De La Chapelle, A. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Rep. 2015, 5, 10566. [Google Scholar] [CrossRef]
- Orois, A.; Gara, S.K.; Mora, M.; Halperin, I.; Martínez, S.; Alfayate, R.; Kebebew, E.; Oriola, J. NOP53 as A Candidate Modifier Locus for Familial Non-Medullary Thyroid Cancer. Genes 2019, 10, 899. [Google Scholar] [CrossRef] [Green Version]
- Gara, S.K.; Jia, L.; Merino, M.J.; Agarwal, S.K.; Zhang, L.; Cam, M.; Patel, D.; Kebebew, E. Germline HABP2 Mutation Causing Familial Nonmedullary Thyroid Cancer. N. Engl. J. Med. 2015, 373, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Tomsic, J.; He, H.; de la Chapelle, A. HABP2 Mutation and Nonmedullary Thyroid Cancer. N. Engl. J. Med. 2015, 373, 2086. [Google Scholar] [CrossRef]
- Tomsic, J.; Fultz, R.; Liyanarachchi, S.; He, H.; Senter, L.; De La Chapelle, A. HABP2 G534E Variant in Papillary Thyroid Carcinoma. PLoS ONE 2016, 11, e0146315. [Google Scholar] [CrossRef] [Green Version]
- Kowalik, A.; Gąsior-Perczak, D.; Gromek, M.; Siołek, M.; Walczyk, A.; Pałyga, I.; Chłopek, M.; Kopczyński, J.; Mężyk, R.; Kowalska, A.; et al. The p.G534E variant of HABP2 is not associated with sporadic papillary thyroid carcinoma in a Polish population. Oncotarget 2017, 8, 58304–58308. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wu, K.; Lin, Z.; Bai, S.; Wu, J.; Li, P.; Xue, H.; Du, J.; Shen, B.; Wang, H.; et al. Identification of susceptibility gene mutations associated with the pathogenesis of familial nonmedullary thyroid cancer. Mol. Genet. Genom. Med. 2019, 7, e1015. [Google Scholar] [CrossRef] [PubMed]
- Sarquis, M.; Moraes, D.C.; Bastos-Rodrigues, L.; Azevedo, P.G.; Ramos, A.V.; Reis, F.V.; Dande, P.V.; Paim, I.; Friedman, E.; De Marco, L. Germline Mutations in Familial Papillary Thyroid Cancer. Endocr. Pathol. 2020, 31, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Siołek, M.; Cybulski, C.; Gąsior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chłopek, M.; Kluźniak, W.; Wokołorczyk, D.; Pałyga, I.; et al. CHEK2mutations and the risk of papillary thyroid cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Incidence of sporadic and familial medullary thyroid cancer (MTC) and non-medullary thyroid cancer (NMTC).
Figure 1.
Incidence of sporadic and familial medullary thyroid cancer (MTC) and non-medullary thyroid cancer (NMTC).


{kind=link}
Table 1.
Genetic alterations in syndromes related to non-medullary thyroid cancer (NMTC).
| Syndrome | Gene | Inheritance Pattern | Other Malignant Tumors | Prevalent Types of Thyroid Tumors | Benign Manifestations | Reference |
|---|---|---|---|---|---|---|
| Ataxia-telangiectasia syndrome * | ATM | AR * | Lymphocytic leukemia, lymphoma, stomach adenocarcinoma, medulloblastoma, glioma | FTC, PTC • | degenerative cerebellar atrophy, telangiectasias, immune defects | [15] |
| ATM | AD | breast cancer, digestive tract cancer, lymphoma, leukemia | [15,16] | |||
| Carney complex | PRKAR1A | AD | - | Follicular hyperplasia, nodular hyperplasia, FA, cystic changes, PTC, FTC | Spotty skin pigmentation (lips, conjunctiva, vaginal, and penile mucosa), cutaneous and mucosal myxoma, cardiac myxoma, breast myxomatosis, primary pigmented nodular adrenocortical disease, GH-producing adenoma, large cell calcifying Sertoli cell tumors, psammomatous melanotic schwannomas | [17] |
| Cowden syndrome | PTEN, SDHB-D, SEC23B, KLLN, PARP4, AKT1, PIK3CA, USF3, TTN, RASAL1 | AD | FTC, breast cancer, epithelial endometrial cancer, colon cancer, renal cell carcinoma melanoma | MNG, Hashimoto thyroiditis, FA, FTC, cPTC, FVPTC, C-cell hyperplasia | Macrocephaly | [18,19] |
| DICER1 syndrome | DICER1 | AD | Pleuropulmonary blastoma, ovarian Sertoli-Leydig cell tumor, genitourinary and cerebral sarcomas | MNG, PTC, FA | MNG, cystic nephroma | [20,21] |
| Familial adenomatous polyposis | APC | AD | Digestive tract cancers, fibrosarcomas | CMVPTC, PTC | Intestinal polyps, osteomas, fibromas, desmoid tumors, dental abnormalities, leiomyomas, congenital hypertrophy of the retinal pigment epithelium | [22,23] |
| Li-Fraumeni syndrome | TP53 | AD | Breast, brain, and adreno cortical cancers and sarcomas | cPTC, FVPTC | [13,24] | |
| Werner syndrome | WRN | AR | Atypical melanoma, bone, or soft tissue sarcomas | FTC, PTC, ATC | Aging, bilateral cataract, type 2 diabetes mellitus, hypogonadism, meningioma | [2,25] |
* Ataxia-telangiectasia syndrome occurs only in autosomal recessive pattern. However, heterozygotic carriers have an increased risk to cancer radio ionizing-induced. • An increased risk for thyroid cancer was observed in relatives of A-T patients, but the histological type was not specified in those epidemiological analysis. The above information is inferred from susceptibility thyroid cancer studies [26,27].
Table 2.
Loci and genes associated with non-syndromic familial non-medullary thyroid cancer (FNMTC).
Table 2.
Loci and genes associated with non-syndromic familial non-medullary thyroid cancer (FNMTC).
| Loci/Gene | Localization | Characteristics | Reference |
|---|---|---|---|
| Linkage analysis | |||
| TCO | 19p13.2 | Oxyphilic PTC | [6] |
| NMTC1 | 2q21 | [9] | |
| PRN1 | 1q21 | Papillary renal cancer | [8] |
| MNG1/DICER1 | 14q32 | MNG | [7] |
| Linkage analysis and NGS | |||
| SRGAP1 | 12q14 | [88] | |
| 8p23.1–p22 | [10] | ||
| 6q22 | [89] | ||
| lncRNA inside TG | 8q24 | Melanoma in 1 family | [90] |
| Enhancer associated with POU2F1 and YY1 | 4q32 | [91] | |
| Other methodology | |||
| NKX2-1 | 14q13.3 | [92] | |
Table 3.
Genes associated with genetic predisposition of sporadic papillary thyroid cancer.
| Locus | Nearest Gene | Population | Reference |
|---|---|---|---|
| 9q22.33 | FOXE1, PTCSC2 | Belarus, Iceland, Italy, Korea, Netherlands Poland, Spain, USA | [98,99,100] |
| 14q13.3 | PTCSC3, NKX2-1, MBIP1 | Iceland, Italy, Korea, Netherlands, Poland, Spain, USA | [98,99] |
| 2q35 | DIRC3 | Iceland, Italy, Korea, Netherlands, Poland, Spain, UK, USA | [99,101] |
| 8p12 | NRG1 | Iceland, Korea, Netherlands, Spain, USA | [99,102] |
| 1q42.2 | PCNXL2 | Iceland, Korea, Netherlands, Spain, USA | [102,103] |
| European Only | |||
| 3q26.2 | LRRC34 | Iceland, Netherlands, Spain, USA | [103] |
| 5p15.33 | TERT | Iceland, Netherlands, Spain, USA | [103] |
| 5q22.1 | EPB41L4A | Iceland, Netherlands, Spain, USA | [103] |
| 10q24.33 | OBFC1 | Iceland, Netherlands, Spain, USA | [103] |
| 15q22.33 | SMAD3 | Iceland, Netherlands, Spain, USA | [103] |
| Korean Only | |||
| 12q14.3 | MSRB3 | Korea | [102] |
| 1p13.3 | VAV3 | Korea | [102] |
| 4q21.1 | SEPT11 | Korea | [102] |
| 3p14.2 | FHIT | Korea | [102] |
| 19p13.2 | INSR | Korea | [102] |
| 12q24.13 | SLC8B1 | Korea | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Miasaki, F.Y.; Fuziwara, C.S.; Carvalho, G.A.d.; Kimura, E.T. Genetic Mutations and Variants in the Susceptibility of Familial Non-Medullary Thyroid Cancer. Genes 2020, 11, 1364. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111364
AMA Style
Miasaki FY, Fuziwara CS, Carvalho GAd, Kimura ET. Genetic Mutations and Variants in the Susceptibility of Familial Non-Medullary Thyroid Cancer. Genes. 2020; 11(11):1364. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111364
Chicago/Turabian StyleMiasaki, Fabíola Yukiko, Cesar Seigi Fuziwara, Gisah Amaral de Carvalho, and Edna Teruko Kimura. 2020. "Genetic Mutations and Variants in the Susceptibility of Familial Non-Medullary Thyroid Cancer" Genes 11, no. 11: 1364. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111364
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.