A Tangle of Genomic Aberrations Drives Multiple Myeloma and Correlates with Clinical Aggressiveness of the Disease: A Comprehensive Review from a Biological Perspective to Clinical Trial Results

 , , and
, , and
Abstract
:1. Introduction
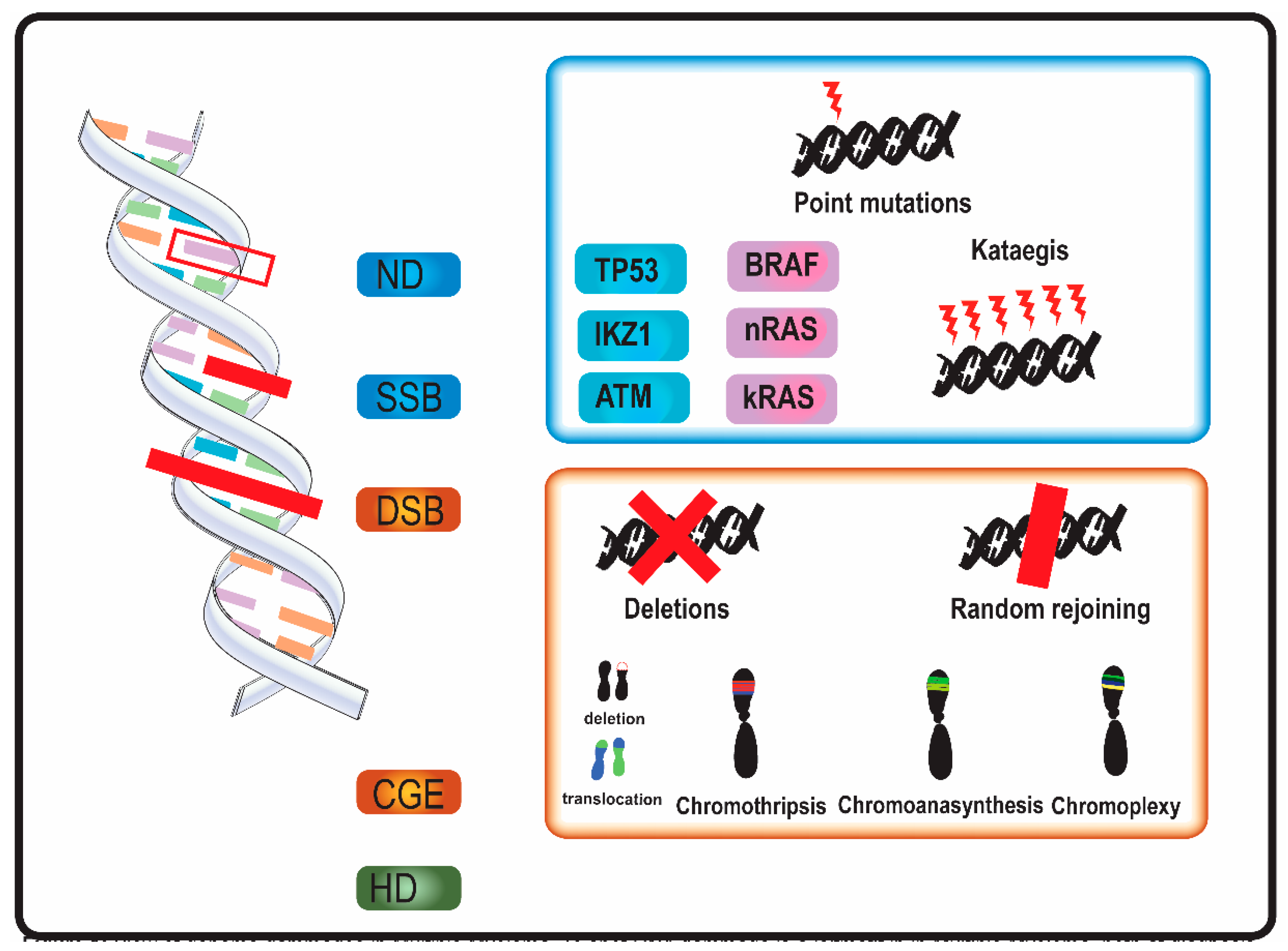
2. Landscape of Genomic Damage in MM
2.1. Timing of Genome Aberrations in MM
2.2. Mechanisms of DNA Damage in MM
2.2.1. Single-Nucleotide Variants and Indels
2.2.2. Copy Number Variations
2.2.3. Chromosomal Translocations
2.2.4. Mutation Clusters
2.2.5. Complex Genome Events
3. Clinical Implications of Genome Aberrations in MM
3.1. Impact on Prognosis of Genome Aberrations and Clinical Trials Results
3.2. Drug-Resistance
3.3. Drug Targeting
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispensieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispensieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak, V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. 2018, 78, 2449–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; de Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.T.; Mudie, L.; et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2018, 32, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Coffey, D.G.; Wu, Q.V.; Towlerton, A.M.H.; Ornelas, S.; Morales, A.J.; Xu, Y.; Green, D.J.; Warren, E.H. Ultradeep, targeted sequencing reveals distinct mutations in blood compared to matched bone marrow among patients with multiple myeloma. Blood Cancer J. 2019, 9, 1–4. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Cordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouelette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustad, E.H.; Yellapantula, V.D.; Glodzik, D.; Maclachlan, K.H.; Diamond, B.; Boyle, E.M.; Ashby, C.; Blaney, P.; Gundem, G.; Hultcrantz, M.; et al. Revealing the Impact of Structural Variants in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 258–273. [Google Scholar] [CrossRef]
- Berry, N.K.; Dixon-McIver, A.; Scott, R.J.; Rowlings, P.; Enjeti, A.K. Detection of complex genomic signatures associated with risk in plasma cell disorders. Cancer Genet. 2017, 218–219, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzales, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Gupta, R.; Mathur, N.; Rani, L.; Kumar, L.; Sharma, A.; Singh, V.; Gupta, A.; Sharma, O.D. Clinical impact of chromothriptic complex chromosomal rearrangements in newly diagnosed multiple myeloma. Leuk. Res. 2019, 76, 58–64. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Magrangeas, F.; Avet-Loiseau, H.; Munshi, N.C.; Minvielle, S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011, 118, 675–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J.; Lee, K.H.; Yoon, K.A.; Sohn, J.Y.; Lee, E.; Lee, H.; Eom, H.S.; Kong, S.Y. Chromothripsis in Treatment Resistance in Multiple Myeloma. Genomics Inform. 2017, 15, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxe, D.; Seo, E.J.; Beaulieu Bergeron, M.; Han, J.Y. Recent advances in cytogenetic characterization of multiple myeloma. Int. J. Lab. Hematol. 2019, 41, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J.; Kumar, S.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mullingan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktas Samur, A.; Minvielle, S.; Shammas, M.; Fulciniti, M.; Magrangeas, F.; Richardson, P.G.; Moreau, P.; Attal, M.; Anderson, K.C.; Parmigiani, G.; et al. Deciphering the chronology of copy number alterations in Multiple Myeloma. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van de Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and targeting of TP53 pathway in multiple myeloma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-Dependent Activation of a MYC Transgene Induces Multiple Myeloma in a Conditional Mouse Model of Post-Germinal Center Malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef] [Green Version]
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzales, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.P.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosom. Cancer 2011, 50, 765–774. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Lionetti, M.; Barbieri, M.; Manzoni, M.; Fabris, S.; Bandini, C.; Todoerti, K.; Nozza, F.; Rossi, D.; Musto, P.; Bandini, L.; et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: An italian cohort study and overview of the literature. Oncotarget 2016, 7, 21353–21361. [Google Scholar] [CrossRef] [Green Version]
- Chin, M.; Sive, J.I.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. 2017, 7, e610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corre, J.; Cleynen, A.; Robiou du Pont, S.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Poot, M. Genes, proteins, and biological pathways preventing chromothripsis. Methods Mol. Biol. 2018, 1769, 231–251. [Google Scholar] [CrossRef] [PubMed]
- Berry, N.K.; Scott, R.J.; Rowlings, P.; Enjeti, A.K. Clinical use of SNP-microarrays for the detection of genome-wide changes in haematological malignancies. Crit. Rev. Oncol. Hematol. 2019, 142, 58–67. [Google Scholar] [CrossRef]
- Sawyer, J.R. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011, 204, 3–12. [Google Scholar] [CrossRef]
- Pellestor, F.; Gatinois, V. Chromoanagenesis: A piece of the macroevolution scenario. Mol. Cytogenet. 2020, 13, 1–9. [Google Scholar] [CrossRef]
- Onodera, N.; McCabe, N.R.; Rubin, C.M. Formation of a hyperdiploid karyotype in childhood acute lymphoblastic leukemia. Blood 1992, 80, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.B.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Petrackova, A.; Minarik, J.; Sedlarikova, L.; Libigerova, T.; Hamplova, A.; Krhovska, P.; Balcarkova, J.; Pika, T.; Papajik, T.; Kriegova, E. Diagnostic deep-targeted next-generation sequencing assessment of TP53 gene mutations in multiple myeloma from the whole bone marrow. Br. J. Haematol. 2020, 189, e122–e125. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [Green Version]
- Tiedemann, R.E.; Gonzalez-Paz, N.; Kyle, R.A.; Santana-Davila, R.; Price-Troska, T.; Van Wier, S.A.; Chng, W.J.; Ketterling, R.P.; Gertz, M.A.; Henderson, K.; et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008, 22, 1044–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avet-Loiseau, H.; Li, C.; Magrangeas, F.; Gourand, W.; Charbonnel, C.; Harousseau, J.L.; Attal, M.; Marit, G.; Mathiot, C.; Facon, T.; et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 2009, 27, 4585–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116. [Google Scholar] [CrossRef] [PubMed]
- Smadja, N.V.; Fruchart, C.; Isnard, F.; Louvet, C.; Dutel, J.L.; Cheron, N.; Grange, M.J.; Monconduit, M.; Bastard, C. Chromosomal analysis in multiple myeloma: Cytogenetic evidence of two different diseases. Leukemia 1998, 12, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Debes-Marun, C.S.; Dewald, G.W.; Bryant, S.; Picken, E.; Santana-Dávila, R.; González-Paz, N.; Winkler, J.M.; Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia 2003, 17, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.J.; Van Wier, S.A.; Ahmann, G.J.; Winkler, J.M.; Jalal, S.M.; Bergsagel, P.L.; Chesi, M.; Trendle, M.C.; Oken, M.M.; Blood, E.; et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood 2005, 106, 2156–2161. [Google Scholar] [CrossRef] [Green Version]
- Avet-Loiseau, H.; Gerson, F.; Magrangeas, F.; Minvielle, S.; Harousseau, J.L.; Bataille, R. Intergroupe Francophone du Myélome. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001, 98, 3082–3086. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Brioli, A.; Boyle, E.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014, 4, e191. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Butrym, A.; Łacina, P.; Rybka, J.; Chaszczewska-Markowska, M.; Mazur, G.; Bogunia-Kubik, K. Cereblon and IRF4 Variants Affect Risk and Response to Treatment in Multiple Myeloma. Arch. Immunol. Ther. Exp. (Warsz) 2016, 64, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.A.; Beedie, S.L.; Chau, C.H.; Venzon, D.J.; Gere, S.; Kazandjian, D.; Korde, N.; Mailankody, S.; Landgren, O.; Figg, W.D. Cereblon gene variants and clinical outcome in multiple myeloma patients treated with lenalidomide. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Jenner, M.W.; Li, C.; Gonzalez, D.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; Morgan, G.J. Integration of global SNP-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma. Blood 2006, 108, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, D.R.; Tonon, G.; Huang, Y.; Zhang, Y.; Sinha, R.; Feng, B.; Stewart, J.P.; Zhan, F.; Khatry, D.; Protopopova, M.; et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell 2006, 9, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, R.; Debes-Marun, C.S.; Picken, E.B.; Dewald, G.W.; Bryant, S.C.; Winkler, J.M.; Blood, E.; Oken, M.M.; Santana-Dávila, R.; González-Paz, N.; et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood 2003, 102, 2562–2567. [Google Scholar] [CrossRef]
- Kaufmann, H.; Ackermann, J.; Baldia, C.; Nösslinger, T.; Wieser, R.; Seidl, S.; Sagaster, V.; Gisslinger, H.; Jäger, U.; Pfeilstöcker, M.; et al. Both IGH translocations and chromosome 13q deletions are early events in monoclonal gammopathy of undetermined significance and do not evolve during transition to multiple myeloma. Leukemia 2004, 18, 1879–1882. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, R.; Bailey, R.J.; Ahmann, G.J.; Rajkumar, S.V.; Hoyer, J.D.; Lust, J.A.; Kyle, R.A.; Gertz, M.A.; Greipp, P.R.; Dewald, G.W. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood 2002, 100, 1417–1424. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Facon, T.; Daviet, A.; Godon, C.; Rapp, M.J.; Harousseau, J.L.; Grosbois, B.; Bataille, R. 14q32 Translocations and Monosomy 13 Observed in Monoclonal Gammopathy of Undetermined Significance Delineate a Multistep Process for the Oncogenesis of Multiple Myeloma 1. CANCER Res. 1999, 59, 4546–4550. [Google Scholar]
- González, D.; Van Der Burg, M.; García-Sanz, R.; Fenton, J.A.; Langerak, A.W.; González, M.; van Dongen, J.J.M.; San Miguel, J.F.; Morgan, G.J. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood 2007, 110, 3112–3121. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J., Jr. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; 1 Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Conticello, S.G. Creative deaminases, self-inflicted damage, and genome evolution. Ann. N. Y. Acad. Sci. 2012, 1267, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Fontana, M.C.; Marconi, G.; Milosevic Feenstra, J.D.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rorà, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, M.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for inference of chromothripsis in cancer genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhurst, S.M. Chromothripsis and telomere crisis: Engines of genome instability. Curr. Opin. Genet. Dev. 2020, 60, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Erez, A.; Sreenath Nagamani, S.C.; Dhar, S.U.; Kołodziejska, K.E.; Dharmadhikari, A.V.; Cooper, M.L.; Wiszniewska, J.; Zhang, F.; Withers, M.A.; et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2011, 146, 889–903. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A.; Carvalho, C.M.B.; Lupski, J.R. A DNA Replication Mechanism for Generating Nonrecurrent Rearrangements Associated with Genomic Disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A Microhomology-Mediated Break-Induced Replication Model for the Origin of Human Copy Number Variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Wilson, T.E.; Glover, T.W. Replication stress and mechanisms of CNV formation. Curr. Opin. Genet. Dev. 2012, 22, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M. Chromoplexy: A New Category of Complex Rearrangements in the Cancer Genome. Cancer Cell 2013, 23, 567–569. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Z.Z.; Leibowitz, M.L.; Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013, 27, 2513–2530. [Google Scholar] [CrossRef] [Green Version]
- Pellestor, F. Chromoanagenesis: Cataclysms behind complex chromosomal rearrangements. Mol. Cytogenet. 2019, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Ni, J.; Liang, Z.; Xue, J.; Fenech, M.F.; Wang, X. The molecular origins and pathophysiological consequences of micronuclei: New insights into an age-old problem. Mutat. Res.-Rev. Mutat. Res. 2019, 779, 1–35. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Tosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Greipp, P.R.; Miguel, J.S.; Durie, B.G.M.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.H.; Davies, F.E.; Pawlyn, C.; Cairns, D.A.; Striha, A.; Collett, C.; Hockaday, A.; Jones, J.R.; Kishore, B.; Garg, M.; et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 57–73. [Google Scholar] [CrossRef] [Green Version]
- Jackson, G.H.; Davies, F.E.; Pawlyn, C.; Cairns, D.A.; Striha, A.; Collett, C.; Waterhouse, A.; Jones, J.R.; Kishore, B.; Garg, M.; et al. Response-adapted intensification with cyclophosphamide, bortezomib, and dexamethasone versus no intensification in patients with newly diagnosed multiple myeloma (Myeloma XI): A multicentre, open-label, randomised, phase 3 trial. Lancet Haematol. 2019, 6, e616–e629. [Google Scholar] [CrossRef] [Green Version]
- Perrot, A.; Lauwers-Cances, V.; Tournay, E.; Hulin, C.; Chretien, M.L.; Royer, B.; Dib, M.; Decaux, O.; Jaccard, A.; Belhadj, K.; et al. Development and validation of a cytogenetic prognostic index predicting survival in multiple myeloma. J. Clin. Oncol. 2019, 37, 1657–1665. [Google Scholar] [CrossRef]
- Samur, M.K.; Aktas Samur, A.; Fulciniti, M.; Szalat, R.; Han, T.; Shammas, M.; Richardson, P.; Magrangeas, F.; Minvielle, S.; Corre, J.; et al. Genome-Wide Somatic Alterations in Multiple Myeloma Reveal a Superior Outcome Group. J. Clin. Oncol. 2020, 38, JCO2000461. [Google Scholar] [CrossRef]
- Yip, K.; Melcher, A.; Harrington, K.; Illidge, T.; Nobes, J.; Webster, A.; Smith, D.; Lorigan, P.; Nathan, P.; Larkin, J. Pembrolizumab in Combination with Radiotherapy for Metastatic Melanoma—Introducing the PERM Trial. Clin. Oncol. 2018, 30, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paner, A.; Patel, P.; Dhakal, B. The evolving role of translocation t(11;14) in the biology, prognosis, and management of multiple myeloma. Blood Rev. 2020, 41. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimaraes, J.E.; Vasconcelos, M.H. Multiple myeloma: Available therapies and causes of drug resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.J.; Dispenzieri, A.; Chim, C.S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Chiecchio, L.; Dagrada, G.P.; Konn, Z.J.; Tapper, W.J.; Walker, B.A.; Wardell, C.P.; Gregory, W.M.; Szubert, A.J.; et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: Analysis of patients treated in the MRC Myeloma IX trial. Leukemia 2012, 26, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, R.; Licht, J.D. MEK and MAF in myeloma therapy. Blood 2011, 117, 2300–2302. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drach, J.; Ackermann, J.; Fritz, E.; Kromer, E.; Schuster, R.; Gisslinger, H.; DeSantis, M.; Zojer, N.; Fiegl, M.; Roka, S.; et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood 1998, 92, 802–809. [Google Scholar] [CrossRef]
- Chang, H.; Qi, C.; Reece, D.; Stewart, A.K. P53 Gene Deletion Detected By Fluorescence in Situ Hybridization Is an Adverse Prognostic Factor for Patients With Multiple Myeloma Following Autologous Stem Cell Transplantation. Blood 2005, 105, 358–360. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase ib study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Oliva, S.; Petrucci, M.T.; Conticello, C.; Catalano, L.; Corradini, P.; Siniscalchi, A.; Magarotto, V.; Pour, L.; Carella, P.; et al. Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma: A randomised, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 1617–1629. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Gay, F.; Schjesvold, F.; Beksac, M.; Hajek, R.; Weisel, K.C.; Goldschmidt, H.; Maisnar, V.; Moreau, P.; Min, C.K.; et al. Oral ixazomib maintenance following autologous stem cell transplantation (TOURMALINE-MM3): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2019, 393, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Cavo, M.; Gay, F.; Beksac, M.; Pantani, L.; Petrucci, M.T.; Dimopoulos, M.A.; Dozza, L.; van der Holt, B.; Zweegman, S.; Oliva, S.; et al. Autologous haematopoietic stem-cell transplantation versus bortezomib–melphalan–prednisone, with or without bortezomib–lenalidomide–dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): A mult. Lancet Haematol. 2020, 7, e456–e468. [Google Scholar] [CrossRef]
- Zweegman, S.; Van Der Holt, B.; Mellqvist, U.H.; Salomo, M.; Bos, G.M.J.; Levin, M.D.; Visser-Wisselaar, H.; Hansson, M.; van der Velden, A.W.G.; Deenik, W.; et al. Melphalan, prednisone, and lenalidomide versus melphalan, prednisone, and thalidomide in untreated multiple myeloma. Blood 2016, 127, 1109–1116. [Google Scholar] [CrossRef] [Green Version]
- Facon, T.; Dimopoulos, M.A.; Dispenzieri, A.; Catalano, J.V.; Belch, A.; Cavo, M.; Pinto, A.; Weisel, K.; Ludwig, H.; Bahlis, N.J.; et al. Final analysis of survival outcomes in the phase 3 FIRST trial of up-front treatment for multiple myeloma. Blood 2018, 131, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Mateos, M.V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Facon, T.; Lee, J.H.; Moreau, P.; Niesvizky, R.; Dimopoulos, M.; Hajek, R.; Pour, L.; Jurczyszyn, A.; Qiu, L.; Klippel, Z.; et al. Carfilzomib or bortezomib with melphalan-prednisone for transplant-ineligible patients with newly diagnosed multiple myeloma. Blood 2019, 133, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Jacobus, S.J.; Cohen, A.D.; Weiss, M.; Callander, N.; Singh, A.K.; Parker, T.L.; Menter, A.; Yang, X.; Parsons, B.; et al. Carfilzomib or bortezomib in combination with lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma without intention for immediate autologous stem-cell transplantation (ENDURANCE): A multicentre, open-label, phase 3, randomise. Lancet Oncol. 2020, 21, 1317–1330. [Google Scholar] [CrossRef]


{kind=link}
| Aberration | Incidence at Diagnosis | Prevalence | Citations |
|---|---|---|---|
| Single nucleotide variants | |||
| MAPK pathway (mostly kRAS, nRAS, BRAF) | 40% | [10,11,38] | |
| NF-KB pathway | 20% | [10,11,38] | |
| DNA-repair pathway | 15% | [38] | |
| TP53 mut | 2–4% | 20–40% | [27,38,39,40,41] |
| Chromosome-level events | |||
| 1q gain | 31–37% | [24,42,43] | |
| +15 | 37% | [43] | |
| +9 | 36% | [43] | |
| +19 | 33% | [43] | |
| +5 | 33% | [43] | |
| +3 | 33% | [43] | |
| +11 | 25% | [43] | |
| +7 | 21% | [43] | |
| 6p gain | 15% | [42,43] | |
| +21 | 12% | [43] | |
| 11q gain | 10% | [42,43] | |
| +X | 9% | [43] | |
| 13q loss | 59–60% | [24,43] | |
| 16q loss | 28–35% | [42,43] | |
| 1p loss | 24–30% | [42,43] | |
| 14q loss | 23–39% | [24,42,43] | |
| 8p loss | 22–25% | [42,43] | |
| 6q loss | 21–33% | [42,43] | |
| −22 | 18% | [43] | |
| 16p loss | 15% | [42] | |
| 12p loss | 13–15% | [42,43] | |
| −20 | 12% | [43] | |
| del(17p) | 7–10% | 80% | [40,41,43] |
| Hyperdyploidy | |||
| Hyperdyploidy | 50% | [19,24,44,45,46] | |
| Translocations | |||
| t(11;14)(q13;q32) IGH/CCND1 | 15% | [19] | |
| t(14;16)(q32;q23) IGH/MAF | 5% | [19] | |
| t(6;14)(p21;q32) IGH/CCND3 | 1–2% | [19] | |
| t(14;20)(q32;q12) IGH/MAFB | 1% | [19] | |
| translocations involving MYC | 15–20% | [19,47,48] | |
| Rare and complex events | |||
| Kataegis | 3% | [19] | |
| Chromothripsis | 1–24% | [15,18,20] | |
| Chromoanasynthesis | Anecdotical-19% | Anecdotical | [15,16,22] |
| Chromoplexy | 4–11% | 4% | [15,17,18] |
| Standard Risk | High Risk | Ultra-High Risk |
|---|---|---|
| Patients not in high risk or ultra-high risk group | t(11;14)(q13;q32) IGH/CCND1 | More than 1 high risk lesion § |
| t(14;16)(q32;q23) IGH/MAF | ||
| t(14;20)(q32;q12) IGH/MAFB | ||
| del (17p) | ||
| gain (1q) § |
| ID | Author (Year) Study Median f-up | Genomic Aberration | Treatment | Number of Patients with Genomic Aberration | MRD- n (%) | sCR, n (%) | CR, n (%) | ORR, n (%) | Median PFS (m) | Median OS (m) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F.M. Gay (2015) [103] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | CRD | 30 (23%) | NA | NA | NA | NA | 25 m (CI NA) * | NR |
| median f-up 52 m | M200-ASCT | 23 (18%) | NA | NA | NA | NA | 33 m (CI NA) * | 58 m (CI NA) | ||
| t(4;14) | CRD | 17 (13%) | NA | NA | NA | NA | NA | NA | ||
| M200-ASCT | 11 (9%) | NA | NA | NA | NA | NA | NA | |||
| t(14;16) | CRD | 6 (5%) | NA | NA | NA | NA | NA | NA | ||
| M200-ASCT | 6 (5%) | NA | NA | NA | NA | NA | NA | |||
| del(17p) | CRD | 10 (8%) | NA | NA | NA | NA | NA | NA | ||
| M200-ASCT | 6 (5%) | NA | NA | NA | NA | NA | NA | |||
| 2 | G. H. Jacskson (2019)—MYELOMA XI maintenance [82] | high risk as gain (1q), t(4;14), t(14;16), t(14, 20), del(17p) | R maintenance | 102 (22%) | NA | NA | NA | NA | 54 m (CI NA) * | NR |
| median f-up 31 m | observation | 56 (17%) | NA | NA | NA | NA | 24 m (CI NA) * | NR | ||
| ultra-high risk (more than one high risk lesions) | R maintenance | 38 (8%) | NA | NA | NA | NA | 23 m (CI NA) * | 45 m (CI NA) * | ||
| observation | 23 (7%) | NA | NA | NA | NA | 7 m (CI NA) * | 33 m (CI NA) * | |||
| t(4;14) | R maintenance | 42 (9%) | NA | NA | NA | NA | 18 events (43%) * | 12 events (29%) | ||
| observation | 24 (7%) | NA | NA | NA | NA | 24 events (100%) * | 10 events (42%) | |||
| t(14;16) | R maintenance | 8 (2%) | NA | NA | NA | NA | 4 events (50%) | 3 events (37%) | ||
| observation | 6 (2%) | NA | NA | NA | NA | 5 events (83%) | 3 events (50%) | |||
| del(17p) | R maintenance | 25 (5%) | NA | NA | NA | NA | 12 events (48%) | 10 events (40%) | ||
| observation | 13 (4%) | NA | NA | NA | NA | 10 events (77%) | 7 events (54%) | |||
| gain (1q) | R maintenance | 104 (23%) | NA | NA | NA | NA | 43 events (41%) * | 24 events (23%) | ||
| observation | 58 (18%) | NA | NA | NA | NA | 38 events (66%) * | 17 events (29%) | |||
| 3 | G. H. Jacskson (2019)—MYELOMA XI intensification § [83] | high risk as gain (1q), t(4;14), t(14;16), t(14, 20), del(17p) | CVD intensification | 32 (25%) | NA | NA | NA | NA | 31 m (CI NA) * | 9 events (28%) |
| median f-up 30 m | no CVD intensification | 48 (37%) | NA | NA | NA | NA | 17 m (CI NA) * | 11 events (23%) | ||
| ultra-high risk (more than one high risk lesions) | CVD intensification | 18 (14%) | NA | NA | NA | NA | 24 m (CI NA) * | 5 events (28%) | ||
| no CVD intensification | 12 (9%) | NA | NA | NA | NA | 8 m (CI NA) * | 7 events (58%) | |||
| t(4;14) | CVD intensification | 19 (7%) | NA | NA | NA | NA | 12 events (63%) * | 4 events (21%) * | ||
| no CVD intensification | 7 (2%) | NA | NA | NA | NA | 7 events (70%) * | 5 events (50%) * | |||
| t(14;16) | CVD intensification | 2 (1%) | NA | NA | NA | NA | 2 events (100%) | 1 event (50%) | ||
| no CVD intensification | 2 (1%) | NA | NA | NA | NA | 2 events (100%) | 1 event (50%) | |||
| del(17p) | CVD intensification | 5 (2%) | NA | NA | NA | NA | 5 events (71%) * | 2 events (29%) * | ||
| no CVD intensification | 12 (4%) | NA | NA | NA | NA | 12 events (86%) * | 8 events (57%) * | |||
| gain (1q) | CVD intensification | 25 (9%) | NA | NA | NA | NA | 25 events (63%) * | 12 events (30%) | ||
| no CVD intensification | 33 (11%) | NA | NA | NA | NA | 33 events (72%) * | 12 events (26%) | |||
| 4 | P. Moreau (2019)—CASSIOPEIA [104] | high risk as at least one abnormality: del(17p) or t(4;14) | Dara-VTD | 82 (15%) | 49 (60%) | 20 (24%) | NA | NA | 15 events (18%) | NA |
| median f-up 18 m | VTD | 86 (16%) | 38 (44%) | 24 (28%) | NA | NA | 22 events (26%) | NA | ||
| 5 | M.A. Dimopoulos (2019)—TOURMALINE-MM3 [105] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | Ixazomib maintenance | 61 (15%) | NA | NA | NA | NA | 38 events (62%) | NA |
| median f-up 31 m | placebo | 54 (21%) | NA | NA | NA | NA | 38 events (70%) | NA | ||
| 6 | M. Cavo (2020)—EMN02/H095 [106] | high risk as at least one abnormality: t(4;14), t(14;16), del(17p) | ASCT | 135 (25%) | NA | NA | NA | NA | 37.3 m (HR 0.63, 0.46–0.88) * | NR (HR 0.66, 0.45–0.99) * |
| Median f-up 42 m | VMP intensification | 90 (25%) | NA | NA | NA | NA | 20.3 m (HR 0.63, 0.46–0.88) * | 51 m (HR 0.66, 0.45–0.99) * | ||
| single ASCT | 42 (25%) | NA | NA | NA | NA | 27 m (HR 0.59, 0.34–1.03) | 54.7% (HR 0.70, 0.35–1.42) | |||
| double ASCT | 39 (22%) | NA | NA | NA | NA | 46 m (HR 0.59, 0.34–1.03) | 61.3% (HR 0.70, 0.35–1.42) | |||
| del(17p) | ASCT | 64 (11%) | NA | NA | NA | NA | NA | NR (HR 0.48, 0.27–0.86) * | ||
| VMP intensification | 41 (10%) | NA | NA | NA | NA | NA | 47 m (HR 0.48, 0.27–0.86) * | |||
| single ASCT | 22 (13%) | NA | NA | NA | NA | 30 m (HR 0.24, 0.09–0.66) * | 57.1% (HR 0.30, 0.08–1.08) | |||
| double ASCT | 18 (10%) | NA | NA | NA | NA | NR (HR 0.24, 0.09–0.66) * | 80.2% (HR 0.30, 0.08–1.08) | |||
| t(4;14) | ASCT | 63 (11%) | NA | NA | NA | NA | NA | NA | ||
| VMP intensification | 48 (12%) | NA | NA | NA | NA | NA | NA | |||
| single ASCT | 16 (9%) | NA | NA | NA | NA | NA | NA | |||
| double ASCT | 20 (11%) | NA | NA | NA | NA | NA | NA | |||
| t(14;16) | ASCT | 20 (4%) | NA | NA | NA | NA | NA | NA | ||
| VMP intensification | 15 (4%) | NA | NA | NA | NA | NA | NA | |||
| single ASCT | 7 (4%) | NA | NA | NA | NA | NA | NA | |||
| double ASCT | 6 (3%) | NA | NA | NA | NA | NA | NA |
| ID | Author (Year) Study Median f-up | Genomic Aberration | Treatment | Number of Patients with Genomic Aberration | MRD- n (%) | sCR, n (%) | CR, n (%) | ORR, n (%) | Median PFS (m) | Median OS (m) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | S. Zweegman (2016)—HOVON87/NMSG18 [107] | del (17p) | MPT-T | 25 (7.9%) | NA | NA | NA | NA | 20 events (80%) | 11 events (44%) |
| median f-up 36 m | MPR-R | 19 (6%) | NA | NA | NA | NA | 18 events (95%) | 10 events (52%) | ||
| t(4;14) | MPT-T | 21 (6.6%) | NA | NA | NA | NA | 20 events (95%) | 12 events (57%) * | ||
| MPR-R | 19 (6%) | NA | NA | NA | NA | 17 events (89%) | 6 events (32%) * | |||
| 1q21 gain | MPT-T | 64 (20.1%) | NA | NA | NA | NA | 49 events (77%) | 29 events (45%) | ||
| MPR-R | 67 (21.1%) | NA | NA | NA | NA | 51 events (76%) | 23 events (34%) | |||
| 2 | T. Facon (2018)—FIRST [108] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | RD continuous | 43 (17%) | NA | NA | 5 (12%) | 33 (77%) | 8.4 | 29.3 |
| median f-up 67 m | RD18 | 52 (20%) | NA | NA | 7 (13%) | 35 (67%) | 17.5 | 24.3 | ||
| MPT | 47 (19%) | NA | NA | 0 | 32 (68%) | 14.6 | 35.5 | |||
| 3 | M.V. Mateos (2018), M.V. Mateos (2019)—ALCYONE [109,110] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | Dara-VMP | 53 (14.9%) | NA | 10 (19%) | 22 (42%) * | 49 (92.5%) * | NA | NR |
| median f-up 40 m | VMP | 45 (12.9%) | NA | 1 (2%) | 11 (24%) * | 33 (73.3%) * | NA | 39.2 | ||
| 4 | T. Facon (2019)—CLARION [111] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | KMP | 54 (11.3%) | NA | NA | NA | NA | ns between arms; HR 0.96 (95% CI: 0.58–1.58) | NA |
| median f-up 27 m | VMP | 67 (14%) | NA | NA | NA | NA | NA | |||
| 5 | G. H. Jacskson (2019)—MYELOMA XI maintenance [82] | high risk as gain (1q), t(4;14), t(14;16), t(14, 20), del(17p) | R maintenance | 64 (27%) | NA | NA | NA | NA | 16 m (CI NA) * | 49 m (CI NA) |
| median f-up 31 m | observation | 57 (28%) | NA | NA | NA | NA | 8 m (CI NA) * | 46 m (CI NA) | ||
| ultra-high risk (more than one high risk lesions) | R maintenance | 15 (6%) | NA | NA | NA | NA | 14 m (CI NA) | 46 m (CI NA) | ||
| observation | 7 (3%) | NA | NA | NA | NA | 11 m (CI NA) | 39 m (CI NA) | |||
| t(4;14) | R maintenance | 9 (4%) | NA | NA | NA | NA | 7 events (78%) | 3 events (34%) | ||
| observation | 8 (4%) | NA | NA | NA | NA | 8 events (100%) | 5 events (62%) | |||
| t(14;16) | R maintenance | 13 (5%) | NA | NA | NA | NA | 11 events (85%) | 8 events (62%) | ||
| observation | 2 (1%) | NA | NA | NA | NA | 1 event (50%) | 1 event (50%) | |||
| del(17p) | R maintenance | 12 (5%) | NA | NA | NA | NA | 8 events (67%) | 5 events (42%) | ||
| observation | 11 (5%) | NA | NA | NA | NA | 11 events (100%) | 7 events (64%) | |||
| gain (1q) | R maintenance | 58 (24) | NA | NA | NA | NA | 60 events (62%) * | 27 events (47%) | ||
| observation | 49 (24%) | NA | NA | NA | NA | 46 events (94%) * | 23 events (47%) | |||
| 6 | S.Z. Usmani (2019)—KEYNOTE-185 [100] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | Pembro-RD | 24 (16%) | NA | NA | NA | NA | NA | 4 events (21%) |
| median f-up: 6.6 m | RD | 10 (7%) | NA | NA | NA | NA | NA | 0 events | ||
| 7 | P.M. Voorhees (2020)—GRIFFIN [112] | high risk cytogenetic as at least one abnormality: del(17p), t(4;14), or t(14;16) | RVd | 14 (14.4%) | 53 (51%) | 42 (42.4%) | 9 (9.1%) | 98 (99%) | NR | NR |
| median f-up 22 m | Dara-RVd | 16 (16.3%) | 21 (20.4%) | 31 (32%) | 10 (10.3%) | 89 (91.8%) | NR | NR | ||
| 8 | S.K. Kumar (2020)—ENDURANCE [113] | t(4;14) | VRd | 44 (12%) | NA | NA | NA | NA | 18.3 m (14.9-NR) | NA |
| median f-up 9 m | KRd | 36 (9%) | NA | NA | NA | NA | 19.3 m (12.7-NR) | NA |
| Patient Population | Recommendations | Evidences for Recommendations |
|---|---|---|
| Transplant eligible patients | No clear advantage has been demonstrated in adding daratumumab in first line therapy to bortezomib-based regimens in high risk patients. | Median PFS 26% VTD vs. 18% Dara-VTD [104] |
| CVD intensification after induction therapy should be offered to high and ultra-high cytogenetic risk patients, including patients with gain 1q and t(14;20), not achieving at least a VGPR before proceeding to ASCT. | Median PFS 31 months with CVD intensification vs. 17 months without CVD intensification [83] | |
| Melphalan 200 mg/sqm followed by ASCT should be preferred to consolidation with CRD in high cytogenetic risk patients. | Median PFS 25 months CRD vs. 33 months M200-ASCT [103] | |
| ASCT should be offered in high cytogenetic risk patients, particularly in del(17p). Furthermore, double ASCT should be preferred to single ASCT in del(17p). | Median PFS 37 m with ASCT vs. 20 m with VMP (HR 0.63, 0.46–0.88). Median OS not reached with ASCT vs. 52 m with VMP (HR 0.66, 0.45–0.99). Median PFS not reached with double ASCT vs. 30 m with single ASCT in del(17p) patients (HR 0.24, 0.09–0.66) [106] | |
| Lenalidomide maintenance should be offered to high and ultra-high cytogenetic risk patients, including patients with gain (1q) and t(14;20). | Median PFS 54 months with R maintenance vs. 24 months with observation) [82] | |
| Transplant ineligible patients | Lenalidomide based-regimen should be preferred in t(4;14) patients. | Better OS; 6 deaths with MPR-R vs. 12 deaths with MPT-T [107] |
| Daratumumab should be added in first line therapy to bortezomib-based triplets in patients with high cytogenetic risk. | ORR 92.5% in Dara-VMP vs. 73.3% in VMP [109,110] | |
| Lenalidomide maintenance should be offered to high cytogenetic risk patients, including patients with gain (1q) and t(14;20), but benefit is not clear in ultra-high risk patients. | Median PFS 16 months with R maintenance vs. 8 months with observation [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sessa, M.; Cavazzini, F.; Cavallari, M.; Rigolin, G.M.; Cuneo, A. A Tangle of Genomic Aberrations Drives Multiple Myeloma and Correlates with Clinical Aggressiveness of the Disease: A Comprehensive Review from a Biological Perspective to Clinical Trial Results. Genes 2020, 11, 1453. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121453
Sessa M, Cavazzini F, Cavallari M, Rigolin GM, Cuneo A. A Tangle of Genomic Aberrations Drives Multiple Myeloma and Correlates with Clinical Aggressiveness of the Disease: A Comprehensive Review from a Biological Perspective to Clinical Trial Results. Genes. 2020; 11(12):1453. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121453
Chicago/Turabian StyleSessa, Mariarosaria, Francesco Cavazzini, Maurizio Cavallari, Gian Matteo Rigolin, and Antonio Cuneo. 2020. "A Tangle of Genomic Aberrations Drives Multiple Myeloma and Correlates with Clinical Aggressiveness of the Disease: A Comprehensive Review from a Biological Perspective to Clinical Trial Results" Genes 11, no. 12: 1453. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121453