Characteristics of Microsatellites Mined from Transcriptome Data and the Development of Novel Markers in Paeonia lactiflora


Abstract
:1. Introduction
2. Materials and Methods
2.1. SSR Identification, Annotation from Transcriptome Data and SSR Primer Design
2.2. Plant Materials
2.3. SSR Primer Evaluation in Eight Cultivars
2.4. Phylogenetic Analysis of Seven Species of Paeonia and 24 Cultivars of P. lactiflora
3. Results
3.1. Numbers and Distribution of SSRs in Transcriptome Data
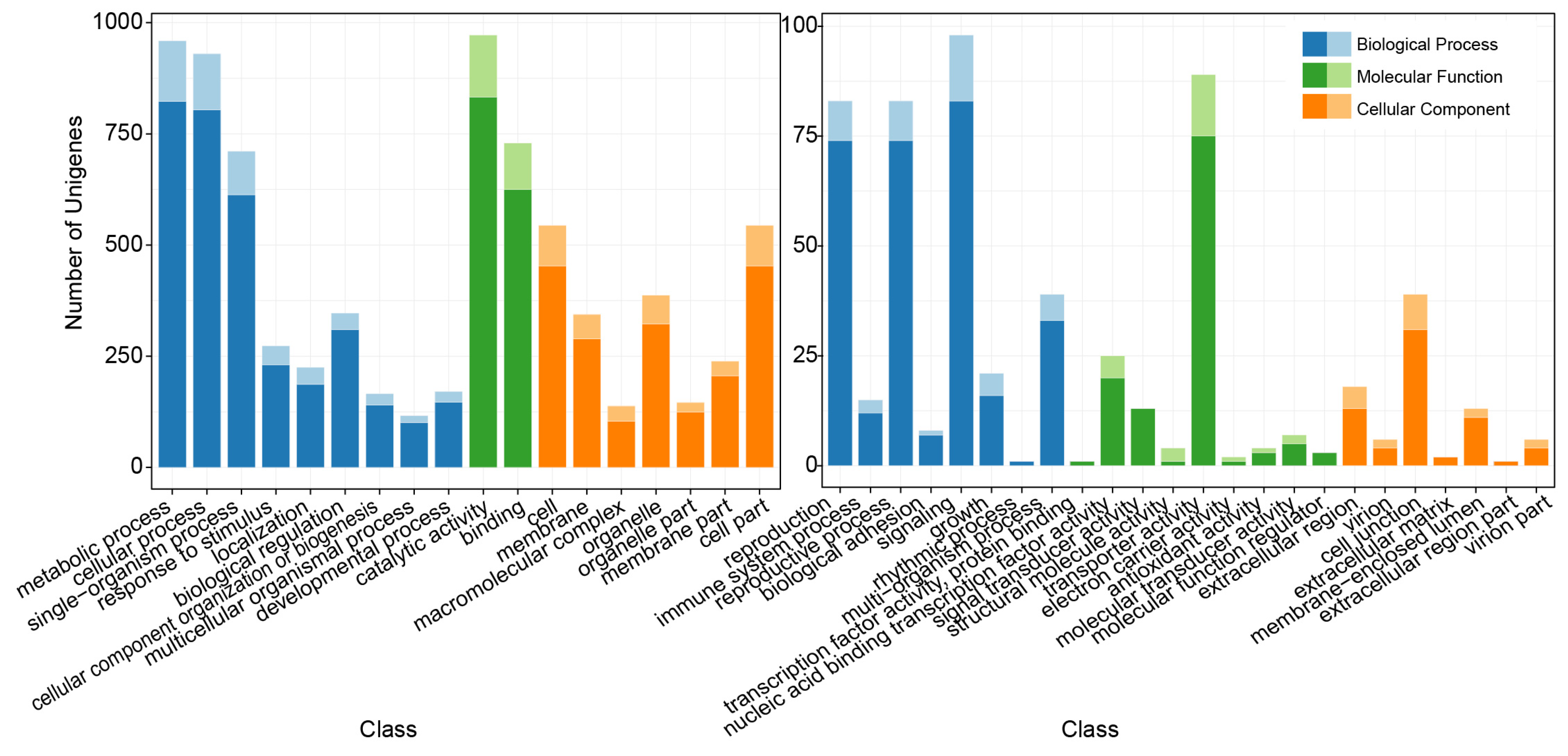
3.2. Annotation of the Unigenes with SSRs
3.3. Initial Amplification of SSR Primers
3.4. Polymorphism in P. lactiflora
3.5. Transferability of SSR Markers among Paeonia Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kamenetsky, R.; Dole, J. Herbaceous peony (Paeonia): Genetics, physiology and cut flower production. Floric. Ornam. Biotechnol. 2012, 6, 62–77. [Google Scholar]
- Collard, B.C.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.; Shrivastava, N.; Padh, H. Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 2008, 27, 617–631. [Google Scholar] [CrossRef]
- Bhattarai, G.; Mehlenbacher, S.A. In silico development and characterization of tri-nucleotide simple sequence repeat markers in hazelnut (Corylus avellana L.). PLoS ONE 2017, 12, e0178061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, G.; Tang, L.; Zhang, Y.; Huang, L.; Ma, X.; Cao, X.; Pan, L.; Zhang, X.; Zhang, X. Development of SSR markers based on transcriptome sequencing and association analysis with drought tolerance in perennial grass Miscanthus from China. Front. Plant Sci. 2017, 8, 801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; An, Y.; Li, F.; Li, S.; Liu, L.; Zhou, Q.; Zhao, S.; Wei, C. Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers for genetic studies in tea plant (Camellia sinensis). Mol. Breed. 2018, 38, 59. [Google Scholar] [CrossRef]
- Taheri, S.; Abdullah, T.L.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 2018, 23, 399. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Kim, C.-H.; Shin, D.-I.; Kim, S.-M.; Koo, H.-M.; Park, Y.-J. Development of simple sequence repeat (SSR) markers to study diversity in the herbaceous peony (Paeonia lactiflora). J. Med. Plants Res. 2011, 5, 6744–6751. [Google Scholar] [CrossRef]
- Li, L.; Cheng, F.Y.; Zhang, Q.X. Microsatellite markers for the Chinese herbaceous peony Paeonia lactiflora (Paeoniaceae). Am. J. Bot. 2011, 98, e16–e18. [Google Scholar] [CrossRef]
- Sun, J.; Yuan, J.; Wang, B.; Pan, J.; Zhang, D. Development and characterization of 10 microsatellite loci in Paeonia lactiflora (Paeoniaceae). Am. J. Bot. 2011, 98, e242–e243. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, B.; Bassil, N.; Nyberg, A.; Knaus, B.; Smith, D.; Barney, D.L.; Hummer, K. Microsatellite marker development in peony using next generation sequencing. J. Am. Soc. Hortic. Sci. 2013, 138, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, Y.; Li, D.; Wang, G.; Li, X.; Xia, Y. Transcriptomic analysis of the underground renewal buds during dormancy transition and release in ‘Hangbaishao’Peony (Paeonia lactiflora). PLoS ONE 2015, 10, e0119118. [Google Scholar]
- Ma, Y.; Cui, J.; Lu, X.; Zhang, L.; Chen, Z.; Fei, R.; Sun, X. Transcriptome analysis of two different developmental stages of Paeonia lactiflora seeds. Int. J. Genom. 2017. [Google Scholar] [CrossRef] [Green Version]
- Barbara, T.; PALMA-SILVA, C.; Paggi, G.M.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, A.; Zhang, S.; Xie, Y.; Yan, L. Using the SSR with fluorescent labeling to establish SSR molecular ID code for cultivars of the Chinese herbaceous peony. J. Beijing For. Univ. 2016, 38, 101–109. [Google Scholar]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Gong, C.; Pan, W.; Zhang, D. Development and application of microsatellites in candidate genes related to wood properties in the Chinese white poplar (Populus tomentosa Carr.). DNA Res. 2012, 20, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.K.; Gangadhar, B.H.; Nookaraju, A.; Kumar, S.; Park, S.W. Development of EST-derived SSR markers in pea (Pisum sativum) and their potential utility for genetic mapping and transferability. Plant Breed. 2012, 131, 118–124. [Google Scholar] [CrossRef]
- Woodhead, M.; McCallum, S.; Smith, K.; Cardle, L.; Mazzitelli, L.; Graham, J. Identification, characterisation and mapping of simple sequence repeat (SSR) markers from raspberry root and bud ESTs. Mol. Breed. 2008, 22, 555–563. [Google Scholar] [CrossRef]
- Wu, J.; Cheng, F.; Cai, C.; Zhong, Y.; Jie, X. Association mapping for floral traits in cultivated Paeonia rockii based on SSR markers. Mol. Genet. Genom. 2017, 292, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Guo, L.-L.; Zhang, L.; Zhang, L.-X.; Ma, H.-L.; Guo, D.-L.; Hou, X.-G. Construction of a genetic linkage map in tree peony (Paeonia Sect. Moutan) using simple sequence repeat (SSR) markers. Sci. Hortic. 2017, 219, 294–301. [Google Scholar]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Avvaru, A.K.; Sowpati, D.T.; Mishra, R.K. Patterns of microsatellite distribution across eukaryotic genomes. BMC Genom. 2019, 20, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.D.; Eggler, P.; Seaton, G.; Rossetto, M.; Ablett, E.M.; Lee, L.S.; Henry, R. Analysis of SSRs derived from grape ESTs. Theor. Appl. Genet. 2000, 100, 723–726. [Google Scholar] [CrossRef]
- Wan, Y.; Hong, A.; Zhang, Y.; Liu, Y. Selection and validation of reference genes of Paeonia lactiflora in growth development and light stress. Physiol. Mol. Biol. Plants 2019, 25, 1097. [Google Scholar] [CrossRef]
- Wan, Y.; Zhang, M.; Hong, A.; Lan, X.; Yang, H.; Liu, Y. Transcriptome and weighted correlation network analyses provide insights into inflorescence stem straightness in Paeonia lactiflora. Plant Mol. Biol. 2020, 102, 239–252. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233. [Google Scholar] [CrossRef]
- Lovin, D.D.; Washington, K.O.; deBruyn, B.; Hemme, R.R.; Mori, A.; Epstein, S.R.; Harker, B.W.; Streit, T.G.; Severson, D.W. Genome-based polymorphic microsatellite development and validation in the mosquito Aedes aegypti and application to population genetics in Haiti. BMC Genom. 2009, 10, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-P.; Cheng, F.-Y.; Zhong, Y.; Cai, C.-F.; Wu, J.; Cui, H.-L. Development of simple sequence repeat (SSR) markers from Paeonia ostii to study the genetic relationships among tree peonies (Paeoniaceae). Sci. Hortic. 2013, 164, 58–64. [Google Scholar] [CrossRef]
- Ji, L.; da Silva, J.A.T.; Zhang, J.; Tang, Z.; Yu, X. Development and application of 15 novel polymorphic microsatellite markers for sect. Paeonia (Paeonia L.). Biochem. Syst. Ecol. 2014, 54, 257–266. [Google Scholar] [CrossRef]
- Kantety, R.V.; La Rota, M.; Matthews, D.E.; Sorrells, M.E. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 2002, 48, 501–510. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Hu, Y.; Zhuang, X.; Xu, W.; Li, P.; Wang, Z. Mining and characterization of novel EST-SSR markers of Parrotia subaequalis (Hamamelidaceae) from the first Illumina-based transcriptome datasets. PLoS ONE 2019, 14, e0215874. [Google Scholar] [CrossRef]
- Park, S.; Son, S.; Shin, M.; Fujii, N.; Hoshino, T.; Park, S. Transcriptome-wide mining, characterization, and development of microsatellite markers in Lychnis kiusiana (Caryophyllaceae). BMC Plant Biol. 2019, 19, 12. [Google Scholar] [CrossRef] [Green Version]
- Bhandawat, A.; Sharma, V.; Singh, P.; Seth, R.; Nag, A.; Kaur, J.; Sharma, R.K. Discovery and utilization of EST-SSR marker resource for genetic diversity and population structure analyses of a subtropical bamboo, Dendrocalamus hamiltonii. Biochem. Genet. 2019, 57, 652–672. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cai, C.; Cheng, F.; Cui, H.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Du, Z.; Ren, J.; Amombo, E.; Hu, T.; Fu, J. Association of SSR markers with functional traits from heat stress in diverse tall fescue accessions. BMC Plant Biol. 2015, 15, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Zhou, L.; Xia, W.; Mason, A.S.; Yang, Y.; Ma, Z.; Peng, M. Exploiting transcriptome data for the development and characterization of gene-based SSR markers related to cold tolerance in oil palm (Elaeis guineensis). BMC Plant Biol. 2014, 14, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosamia, T.C.; Mishra, G.P.; Thankappan, R.; Dobaria, J.R. Novel and stress relevant EST derived SSR markers developed and validated in peanut. PLoS ONE 2015, 10, e0129127. [Google Scholar] [CrossRef] [Green Version]
- Molla, K.A.; Debnath, A.B.; Ganie, S.A.; Mondal, T.K. Identification and analysis of novel salt responsive candidate gene based SSRs (cgSSRs) from rice (Oryza sativa L.). BMC Plant Biol. 2015, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Ruan, X.; Wang, Z.; Su, Y.; Wang, T. Characterization and application of EST-SSR markers developed from the transcriptome of Amentotaxus argotaenia (Taxaceae), a relict vulnerable conifer. Front. Genet. 2019, 10, 1014. [Google Scholar] [CrossRef]
- Song, Q.; Jia, G.; Zhu, Y.; Grant, D.; Nelson, R.T.; Hwang, E.-Y.; Hyten, D.L.; Cregan, P.B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_1. 0) in soybean. Crop Sci. 2010, 50, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, L.; Peng, Z.; Sun, H.; Yue, X.; Lou, Y.; Dong, L.; Wang, L.; Gao, Z. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 2015, 5, 8018. [Google Scholar] [CrossRef]
- Gao, Z.; Wu, J.; Liu, Z.; Wang, L.; Ren, H.; Shu, Q. Rapid microsatellite development for tree peony and its implications. BMC Genom. 2013, 14, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar] [PubMed]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddou-Muratorio, S.; Vendramin, G.G.; Buiteveld, J.; Fady, B. Population estimators or progeny tests: What is the best method to assess null allele frequencies at SSR loci? Conserv. Genet. 2009, 10, 1343. [Google Scholar] [CrossRef]
- Dakin, E.; Avise, J. Microsatellite null alleles in parentage analysis. Heredity 2004, 93, 504–509. [Google Scholar] [CrossRef]
- Molla, K.A.; Azharudheen, T.P.M.; Ray, S.; Sarkar, S.; Swain, A.; Chakraborti, M.; Vijayan, J.; Singh, O.N.; Baig, M.J.; Mukherjee, A.K. Novel biotic stress responsive candidate gene based SSR (cgSSR) markers from rice. Euphytica 2019, 215, 17. [Google Scholar] [CrossRef]
- Hong, D.; Pan, K. A taxonomic revision of the Paeonia anomala complex (Paeoniaceae). Ann. Mo. Bot. Gard. 2004, 91, 87–98. [Google Scholar]
- Hong, D. Peonies of the World: Taxonomy and Phytogeography; Royal Botanic Gardens: Edinburgh, UK, 2010; pp. 33–44. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Repeat Motif | Size Range (bp) | N | Na | Ne | I | Ho | He | Null allele | F | PIC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| T125 | (TC)9 | 128–170 | 31 | 15 | 7.84 | 2.29 | 0.77 | 0.87 | 0.073 | 0.11 | 0.86 |
| T163 | (TA)8 | 146–160 | 31 | 8 | 3.96 | 1.63 | 0.65 | 0.75 | 0.085 | 0.14 | 0.71 |
| T237 | (CT)10 | 95–109 | 31 | 8 | 4.59 | 1.76 | 0.68 | 0.78 | 0.095 | 0.13 | 0.75 |
| T241 | (GA)15 | 127–157 | 31 | 12 | 6.77 | 2.11 | 0.81 | 0.85 | / | 0.05 | 0.84 |
| C160 | (AT)9ctcctt(CTC)5 | 211–227 | 29 | 8 | 4.58 | 1.79 | 0.66 | 0.78 | 0.117 | 0.16 | 0.76 |
| TA564 | (TA)8 | 172–201 | 26 | 9 | 5.43 | 1.90 | 0.77 | 0.82 | 0.058 | 0.06 | 0.79 |
| T317 | (CT)10 | 157–183 | 30 | 9 | 5.94 | 1.92 | 0.83 | 0.83 | / | 0.00 | 0.81 |
| S024 | (AAT)16 | 161–201 | 31 | 10 | 5.60 | 1.94 | 0.77 | 0.82 | 0.042 | 0.06 | 0.80 |
| T040 | (GA)7 | 228–246 | 31 | 7 | 5.02 | 1.73 | 0.68 | 0.80 | 0.106 | 0.15 | 0.77 |
| T179 | (AT)7 | 235–269 | 31 | 10 | 4.93 | 1.83 | 0.71 | 0.80 | 0.070 | 0.11 | 0.77 |
| T192 | (CT)8 | 212–230 | 31 | 8 | 4.50 | 1.75 | 0.68 | 0.78 | 0.093 | 0.13 | 0.75 |
| T300 | (AT)10 | 218–238 | 31 | 9 | 4.45 | 1.80 | 0.48 | 0.78 | 0.171 | 0.38 | 0.75 |
| TA673 | (CA)18 | 190–288 | 31 | 15 | 6.74 | 2.20 | 0.81 | 0.85 | 0.058 | 0.05 | 0.84 |
| T210 | (GA)10 | 253–285 | 31 | 12 | 4.44 | 1.86 | 0.81 | 0.77 | 0.044 | −0.04 | 0.75 |
| TA038 | (CT)9 | 259–299 | 31 | 13 | 6.74 | 2.18 | 0.13 | 0.85 | 0.391 | 0.85 | 0.84 |
| S033 | (TAT)7 | 137–181 | 31 | 10 | 2.47 | 1.39 | 0.42 | 0.59 | 0.123 | 0.29 | 0.57 |
| T304 | (GA)10 | 147–163 | 30 | 9 | 4.75 | 1.77 | 0.63 | 0.79 | 0.144 | 0.20 | 0.76 |
| T863 | (AG)10 | 140–160 | 31 | 10 | 6.26 | 2.03 | 0.74 | 0.84 | 0.070 | 0.12 | 0.82 |
| TA028 | (AC)6 | 101–123 | 31 | 8 | 3.88 | 1.69 | 0.48 | 0.74 | 0.177 | 0.35 | 0.72 |
| T239 | (CT)9 | 185–221 | 31 | 11 | 2.98 | 1.59 | 0.58 | 0.66 | 0.069 | 0.13 | 0.64 |
| TA144 | (CT)6 | 186–286 | 31 | 9 | 4.00 | 1.64 | 0.68 | 0.75 | / | 0.10 | 0.71 |
| T852 | (AT)8 | 158–367 | 31 | 13 | 5.95 | 2.06 | 0.35 | 0.83 | 0.268 | 0.57 | 0.81 |
| F106 | (TATG)5 | 200–290 | 31 | 13 | 5.02 | 2.03 | 0.55 | 0.80 | 0.145 | 0.32 | 0.78 |
| TA082 | (AG)6 | 242–280 | 30 | 13 | 6.00 | 2.03 | 0.73 | 0.83 | 0.084 | 0.12 | 0.81 |
| TA079 | (AT)8 | 232–264 | 29 | 8 | 2.93 | 1.45 | 0.52 | 0.66 | 0.161 | 0.21 | 0.62 |
| T356 | (AG)9 | 247–269 | 30 | 9 | 4.76 | 1.81 | 0.33 | 0.79 | 0.296 | 0.58 | 0.76 |
| TA133 | (AT)8 | 258–286 | 28 | 9 | 4.28 | 1.72 | 0.75 | 0.77 | 0.095 | 0.02 | 0.73 |
| SA010 | (AAT)5 | 264–294 | 31 | 9 | 4.98 | 1.85 | 0.71 | 0.80 | 0.079 | 0.11 | 0.77 |
| W75 | (CTCAC)5 | 257–293 | 31 | 8 | 4.65 | 1.70 | 0.65 | 0.79 | 0.092 | 0.18 | 0.75 |
| T205 | (TC)9 | 275–301 | 31 | 13 | 6.94 | 2.18 | 0.71 | 0.86 | / | 0.17 | 0.84 |
| S853 | (TTG)5 | 158–186 | 29 | 8 | 4.67 | 1.71 | 0.69 | 0.79 | / | 0.12 | 0.75 |
| T859 | (AG)19 | 166–200 | 29 | 11 | 7.61 | 2.15 | 0.90 | 0.87 | 0.000 | -0.03 | 0.85 |
| TA566 | (GA)6 | 167–185 | 25 | 7 | 4.83 | 1.71 | 0.80 | 0.79 | / | −0.01 | 0.76 |
| TA695 | (AC)15 | 164–180 | 30 | 8 | 3.00 | 1.49 | 0.57 | 0.67 | 0.119 | 0.15 | 0.64 |
| S237 | (CTG)8 | 193–219 | 31 | 13 | 9.96 | 2.41 | 0.74 | 0.90 | 0.081 | 0.18 | 0.89 |
| TA464 | (TC)8 | 200–214 | 30 | 8 | 5.56 | 1.85 | 0.73 | 0.82 | 0.062 | 0.11 | 0.80 |
| T160 | (CT)10 | 205–227 | 30 | 10 | 5.47 | 1.92 | 0.70 | 0.82 | / | 0.14 | 0.80 |
| TA134 | (GA)16 | 142–224 | 29 | 13 | 5.43 | 2.09 | 0.93 | 0.82 | 0.019 | −0.14 | 0.80 |
| SA061 | (GAA)7 | 257–277 | 29 | 6 | 3.43 | 1.46 | 0.38 | 0.71 | 0.281 | 0.46 | 0.67 |
| TA074 | (CT)8 | 260–306 | 30 | 13 | 5.79 | 2.03 | 0.97 | 0.83 | 0.000 | −0.17 | 0.81 |
| TA704 | (TC)17 | 237–277 | 30 | 7 | 5.19 | 1.73 | 0.70 | 0.81 | 0.111 | 0.13 | 0.78 |
| TA610 | (TC)8 | 257–291 | 28 | 11 | 6.25 | 2.05 | 0.68 | 0.84 | 0.117 | 0.19 | 0.82 |
| S025 | (ATT)7 | 268–350 | 29 | 13 | 5.14 | 2.02 | 0.55 | 0.81 | 0.200 | 0.32 | 0.79 |
| TA022 | (TC)6 | 276–318 | 30 | 16 | 6.87 | 2.23 | 0.90 | 0.85 | 0.000 | −0.05 | 0.84 |
| TA086 | (AG)10 | 393–419 | 31 | 10 | 5.62 | 1.94 | 0.74 | 0.82 | 0.077 | 0.10 | 0.80 |
| TA700 | (AG)9 | 278–312 | 29 | 13 | 5.76 | 2.05 | 0.62 | 0.83 | 0.155 | 0.25 | 0.81 |
| Mean | 30.07 | 10.26 | 5.26 | 1.88 | 0.67 | 0.80 | 0.11 | 0.16 | 0.77 | ||
| SE | 0.20 | 0.36 | 0.21 | 0.03 | 0.02 | 0.01 | 0.01 | 0.03 | 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Y.; Zhang, M.; Hong, A.; Zhang, Y.; Liu, Y. Characteristics of Microsatellites Mined from Transcriptome Data and the Development of Novel Markers in Paeonia lactiflora. Genes 2020, 11, 214. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020214
Wan Y, Zhang M, Hong A, Zhang Y, Liu Y. Characteristics of Microsatellites Mined from Transcriptome Data and the Development of Novel Markers in Paeonia lactiflora. Genes. 2020; 11(2):214. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020214
Chicago/Turabian StyleWan, Yingling, Min Zhang, Aiying Hong, Yixuan Zhang, and Yan Liu. 2020. "Characteristics of Microsatellites Mined from Transcriptome Data and the Development of Novel Markers in Paeonia lactiflora" Genes 11, no. 2: 214. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020214