Two Novel FAM20C Variants in a Family with Raine Syndrome

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
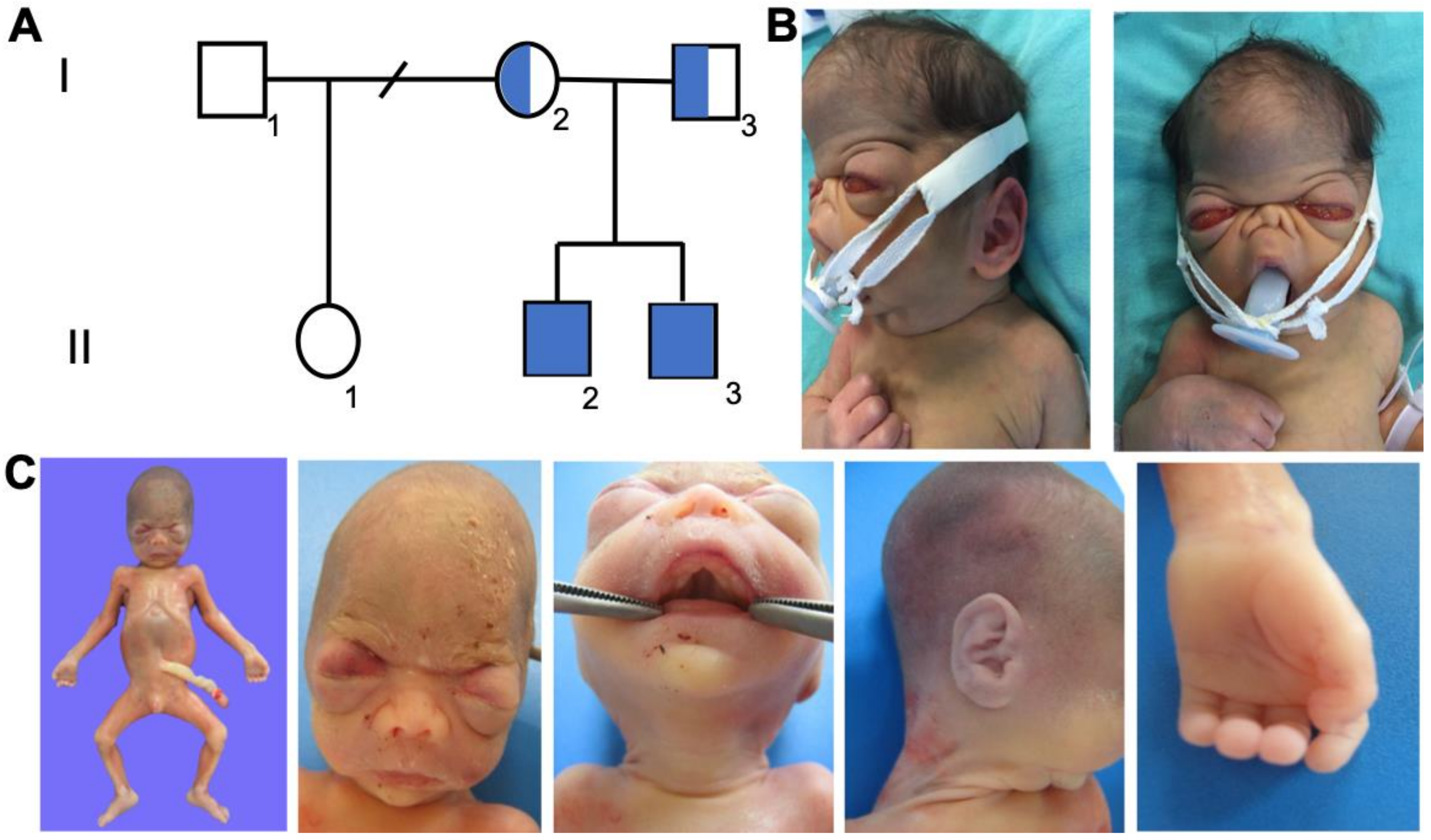
2.1. Cases 1 and 2
2.2. Genetic Analysis
2.3. Immuno-Histochemical Staining for FAM20C
3. Results
3.1. Case Reports
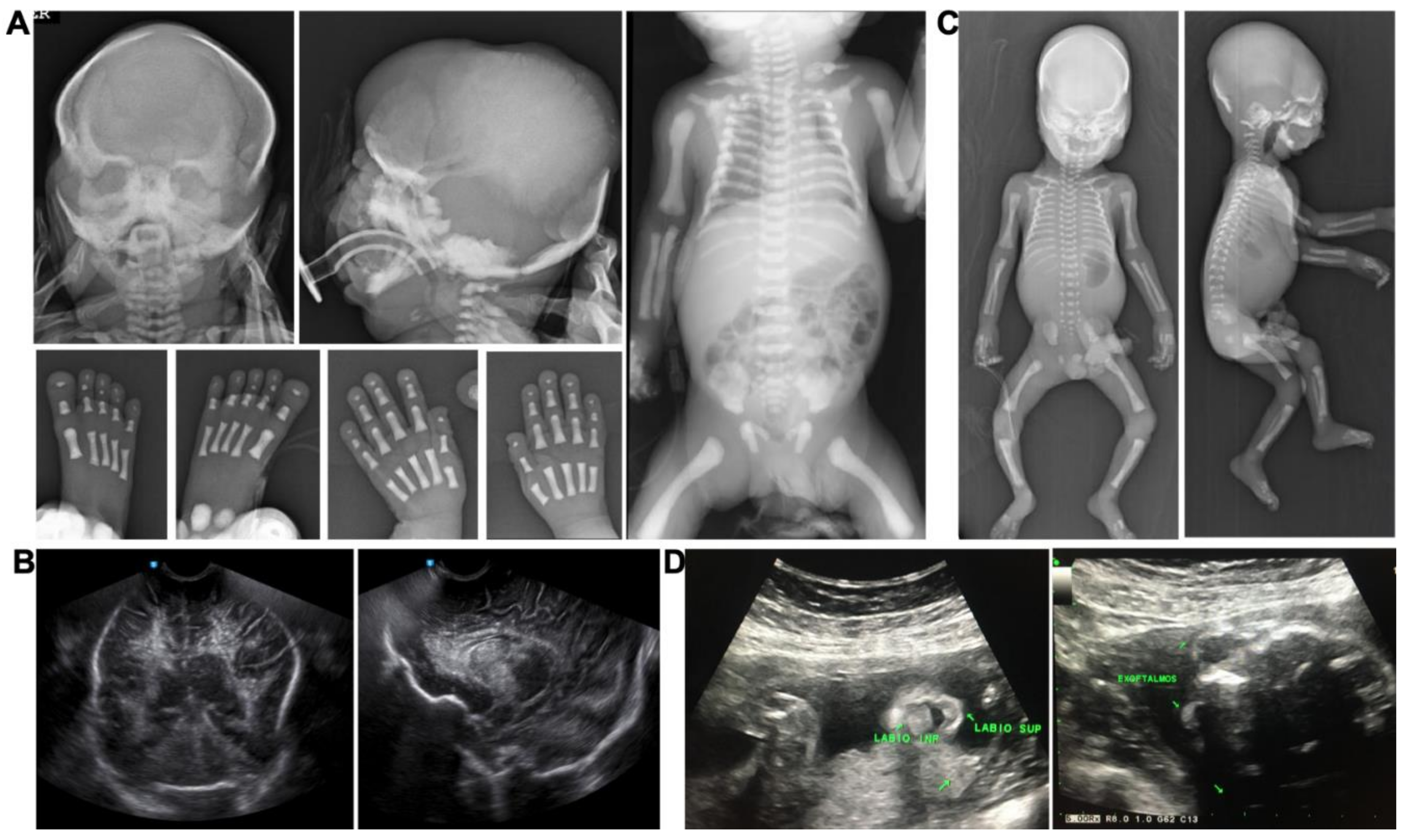
3.2. Pathological Features
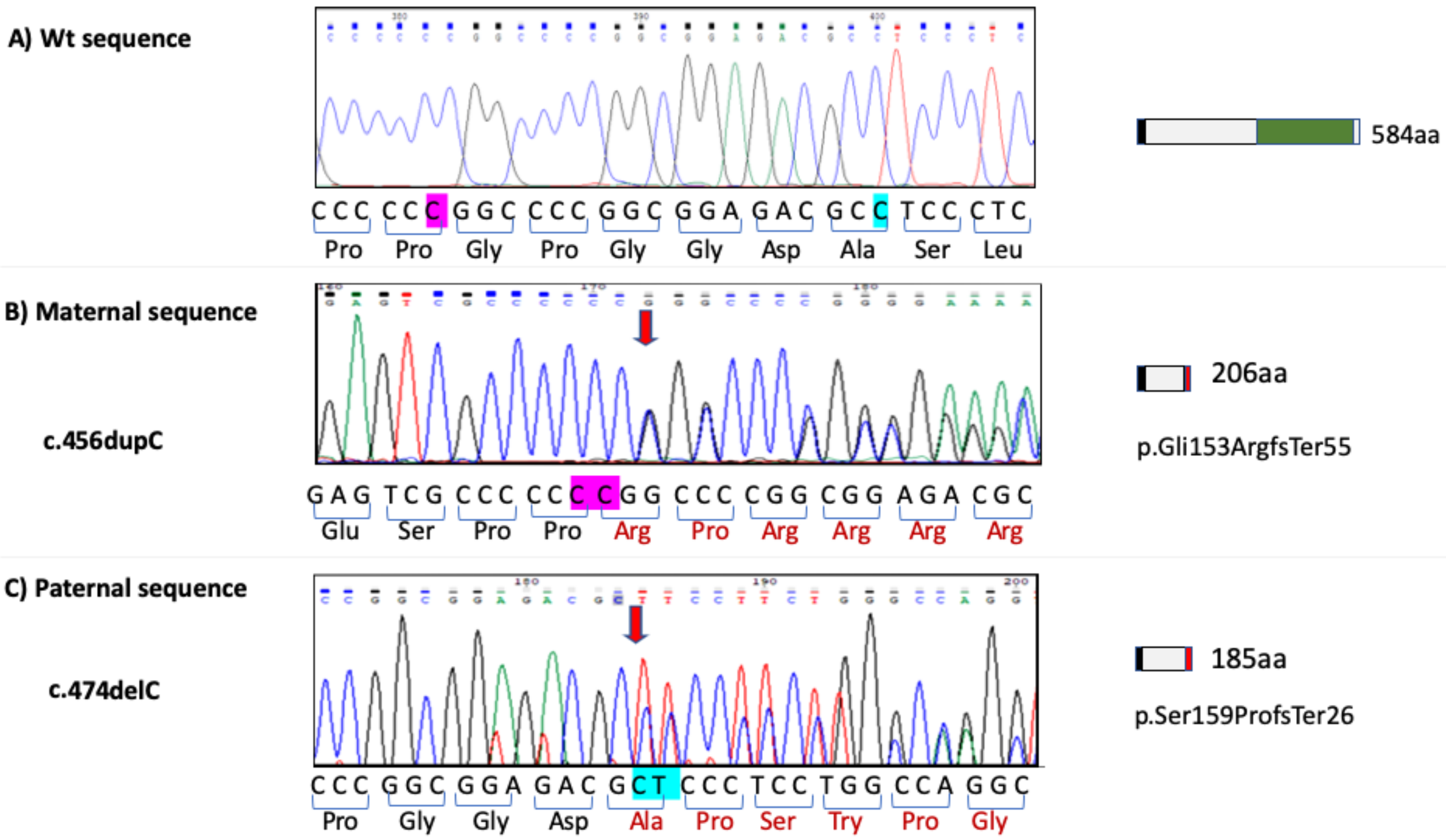
3.3. FAM20C Sequence Analysis
3.4. Immunohistochemical Detection of FAM20C
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raine, J.; Winter, R.M.; Davey, A.; Tucker, S.M. Unknown syndrome: Microcephaly, hypoplastic nose, exophthalmos, gum hyperplasia, cleft palate, low set ears, and osteosclerosis. J. Med. Genet. 1989, 26, 786–788. [Google Scholar] [CrossRef] [Green Version]
- Faundes, V.; Castillo-Taucher, S.; Gonzalez-Hormazabal, P.; Chandler, K.; Crosby, A.; Chioza, B. Raine syndrome: An overview. Eur. J. Med. Genet. Elsevier Masson SAS 2014, 57, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Tada, K.; Takeuchi, A.; Nakamura, M.; Marunaka, H.; Washio, Y.; Tanaka, H.; Miya, F.; Okamoto, N.; Kageyama, M. Fetal ultrasonographic findings including cerebral hyperechogenicity in a patient with non-lethal form of Raine syndrome. Am. J. Med. Genet. Part A 2018, 176, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Rolvien, T.; Kornak, U.; Schinke, T.; Amling, M.; Oheim, R. A novel FAM20C mutation causing hypophosphatemic osteomalacia with osteosclerosis (mild Raine syndrome) in an elderly man with spontaneous osteonecrosis of the knee. Osteoporos. Int. 2019, 30, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Sheth, J.; Bhavsar, R.; Gandhi, A.; Sheth, F.; Pancholi, D. A case of Raine syndrome presenting with facial dysmorphy and review of literature. BMC Med. Genet. 2018, 19. [Google Scholar] [CrossRef]
- Patel, P.J.; Kolawole, T.M.; al-Mofada, S.; Malabarey, T.M.; Hulailah, A. Osteopetrosis: Brain ultrasound and computed tomography findings. Eur. J. Pediatr. 1992, 151, 827–828. [Google Scholar] [CrossRef]
- Rickert, C.H.; Rieder, H.; Rehder, H.; Hülskamp, G.; Hörnig-Franz, I.; Louwen, F.; Paulus, W. Neuropathology of Raine syndrome. Acta Neuropathol. 2002, 103, 281–287. [Google Scholar] [CrossRef]
- Chitayat, D.; Shannon, P.; Keating, S.; Toi, A.; Blaser, S.; Friedberg, T.; Superti-Furga, A.; Chong, K.; Unger, S. Raine syndrome: A rare lethal osteosclerotic bone dysplasia. Prenatal diagnosis, autopsy, and neuropathological findings. Am. J. Med. Genet. Part A. 2007, 143, 3280–3285. [Google Scholar] [CrossRef]
- Michael, K.; Nelson, D.M.; Ortmeier, A. Raine Syndrome. J. Diagn. Med. Sonogr. 2011, 27, 167–170. [Google Scholar] [CrossRef]
- Koob, M.; Doray, B.; Fradin, M.; Astruc, D.; Dietemann, J.L. Raine syndrome: Expanding the radiological spectrum. Pediatr. Radiol. 2011, 41, 389–393. [Google Scholar] [CrossRef]
- Simpson, M.A.; Hsu, R.; Keir, L.S.; Hao, J.; Sivapalan, G.; Ernst, L.M.; Zackai, E.H.; Al-Gazali, L.I.; Hulskamp, G.; Kingston, H.M.; et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am. J. Hum. Genet. 2007, 81, 906–912. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhu, Q.; Cui, J.; Wang, Y.; Chen, M.J.; Guo, X.; Tagliabracci, V.S.; Dixon, J.E.; Xiao, J. Structure and evolution of the Fam20 kinases. Nat. Commun. 2018, 9, 1218. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Tagliabracci, V.S.; Wen, J.; Kim, S.-A.; Dixon, J.E. Crystal structure of the Golgi casein kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10574–10579. [Google Scholar] [CrossRef] [Green Version]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A single kinase generates the majority of the secreted phosphoproteome. Cell 2015, 161, 1619–1632. [Google Scholar] [CrossRef] [Green Version]
- Sreelatha, A.; Kinch, L.N.; Tagliabracci, V.S. The secretory pathway kinases. Biochim. Biophys. Acta 2015, 1854, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, P.; Li, X.; Zhang, S.; Tian, M.; Dai, Y.; Fu, L. Systematic network-based discovery of a Fam20C inhibitor (FL-1607) with apoptosis modulation in triple-negative breast cancer. Mol. Biosyst. 2016, 12, 2108–2118. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Pinna, L.A.; Dixon, J.E. Response to Wang et al.: Secreted protein kinases? Trends Biochem. Sci. 2013, 38, 425. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Jani, P.; Wang, X.; Lu, Y.; Li, N.; Xiao, J.; Qin, C. FAM20C regulates osteoblast behaviors and intracellular signaling pathways in a cell-autonomous manner. J. Cell. Physiol. 2018, 233, 3476–3486. [Google Scholar] [CrossRef]
- Liu, P.; Ma, S.; Zhang, H.; Liu, C.; Lu, Y.; Chen, L.; Qin, C. Specific ablation of mouse Fam20C in cells expressing type I collagen leads to skeletal defects and hypophosphatemia. Sci. Rep. 2017, 7, 3590. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, Y.; Hori, M.; Taguchi, M.; Fukumoto, S. Functional analysis of mutant FAM20C in Raine syndrome with FGF23-related hypophosphatemia. Bone 2014, 67, 145–151. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Appaiah, H.N.; Koller, A.; Nizet, V.; White, K.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.A.; Scheuerle, A.; Hurst, J.; Patton, M.A.; Stewart, C.; Crosby, A.H. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin. Genet. 2009, 75, 271–276. [Google Scholar] [CrossRef]
- Das, D.S.; Patel, T.; Shah, C. Raine syndrome: A rare case presentation. Int. J. Med. Sci. Clin. Invent. 2017, 4, 3304–3306. [Google Scholar] [CrossRef]
- Fradin, M.; Stoetzel, C.; Muller, J.; Koob, M.; Christmann, D.; Debry, C.; Kohler, M.; Isnard, M.; Astruc, D.; Desprez, D.; et al. Osteosclerotic bone dysplasia in siblings with a Fam20C mutation. Clin. Genet. 2011, 80, 177–183. [Google Scholar] [CrossRef]
- Rafaelsen, S.H.; Raeder, H.; Fagerheim, A.K.; Knappskog, P.; Carpenter, T.O.; Johansson, S.; Bjerknes, R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J. Bone Miner. Res. 2013, 28, 1378–1385. [Google Scholar] [CrossRef]
- Mahmood, N.; Donne, A.; Weber, A.; Dharmraj, P. Raine syndrome: A review and a report of metabolic bone disease as a new link. Research 2014, 1, 890. [Google Scholar] [CrossRef]
- Takeyari, S.; Yamamoto, T.; Kinoshita, Y.; Fukumoto, S.; Glorieux, F.H.; Michigami, T.; Hasegawa, K.; Kitaoka, T.; Kubota, T.; Imanishi, Y.; et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone 2014, 67, 56–62. [Google Scholar] [CrossRef]
- Vishwanath, B.; Srinivasa, K.; Veera Shankar, M. Raine syndrome. Indian J. Hum. Genet. 2014, 20, 72–74. [Google Scholar] [CrossRef]
- Acevedo, A.C.; Poulter, J.A.; Alves, P.G.; de Lima, C.L.; Castro, L.C.; Yamaguti, P.M.; Paula, L.M.; Parry, D.A.; Logan, C.V.; Smith, C.E.L.; et al. Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med. Genet. 2015, 16, 8. [Google Scholar] [CrossRef] [Green Version]
- Elalaoui, S.C.; Al-Sheqaih, N.; Ratbi, I.; Urquhart, J.E.; O’Sullivan, J.; Bhaskar, S.; Williams, S.S.; Ellaloussi, M.; Lyahyai, J.; Sbihi, L.; et al. Non lethal Raine syndrome and differential diagnosis. Eur. J. Med. Genet. 2016, 59, 577–583. [Google Scholar] [CrossRef]
- Acosta, A.X.; Peres, L.C.; Chimelli, L.C.; Pina-Neto, J.M. Raine dysplasia: A Brazilian case with a mild radiological involvement. Clin. Dysmorphol. 2000, 9, 99–101. [Google Scholar] [CrossRef]
- Mamedova, E.; Dimitrova, D.; Przhiyalkovskaya, E.; Buryakina, S.; Vasilyev, E.; Tiulpakov, A.; Belaya, Z. Non-lethal Raine syndrome in a middle-aged woman caused by a novel FAM20C mutation. Calcif. Tissue Int. 2019, 105, 567–572. [Google Scholar] [CrossRef]
- Kingston, H.M.; Freeman, J.S.; Hall, C.M. A new lethal sclerosing bone dysplasia. Skeletal. Radiol. 1991, 20, 117–119. [Google Scholar] [CrossRef]
- Kan, A.E.; Kozlowski, K. New distinct lethal osteosclerotic bone dysplasia (Raine syndrome). Am. J. Med. Genet. 1992, 43, 860–864. [Google Scholar] [CrossRef]
- Rejjal, A. Raine syndrome. Am. J. Med. Genet. 1998, 78, 382–385. [Google Scholar] [CrossRef]
- Shalev, S.A.; Shalev, E.; Reich, D.; Borochowitz, Z.U. Osteosclerosis, hypoplastic nose, and proptosis (Raine syndrome): Further delineation. Am. J. Med. Genet. 1999, 86, 274–277. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 2015, 30338. [Google Scholar] [CrossRef] [Green Version]
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Mane, K.; Coates, R.K.; McDonald, P. Intracranial calcification in Raine syndrome. Pediatr. Radiol. 1996, 26, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Al Mane, K.; Al-Dayel, F.; McDonald, P. Intracranial calcification in Raine syndrome: Radiological pathological correlation. Pediatr. Radiol. 1998, 28, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; McAlister, W.H.; Fallon, M.D.; Pierpont, M.E.; Bijanki, V.N.; Duan, S.; Otaify, G.A.; Sly, W.S.; Mumm, S. Raine syndrome (OMIM #259775), caused by FAM20C mutation, is congenital sclerosing osteomalacia with cerebral calcification (OMIM 259660). J. Bone Miner. Res. 2017, 32, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Mahafza, T.; El-Shanti, H.; Omari, H. Raine syndrome: Report of a case with hand and foot anomalies. Clin. Dysmorphol. 2001, 10, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Al-Gazali, L.I.; Jehier, K.; Nazih, B.; Abtin, F.; Haas, D.; Sadagahatian, R. Further delineation of Raine syndrome. Clin. Dysmorphol. 2003, 12, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Hülskamp, G.; Wieczorek, D.; Rieder, H.; Louwen, F.; Hörnig-Franz, I.; Rickert, C.H.; Horst, J.; Harms, E.; Rehder, H. Raine syndrome: Report of a family with three affected sibs and further delineation of the syndrome. Clin. Dysmorphol. 2003, 12, 153–160. [Google Scholar] [CrossRef]
- Kochar, G.S.; Choudhary, A.; Gadodia, A.; Gupta, N.; Simpson, M.A.; Crosby, A.H.; Kabra, M. Raine syndrome: A clinical, radiographic and genetic investigation of a case from the Indian subcontinent. Clin. Dysmorphol. 2010, 19, 153–156. [Google Scholar] [CrossRef]
- Gaigi, S.S.; Zeghal, D.; Masmoudi, A.; Cherif, A.; Jabnoun, S.; Khrouf, N. Raine syndrome. Tunis Med. 2011, 89, 723–725. [Google Scholar]
- Ababneh, F.K.; AlSwaid, A.; Youssef, T.; Al Azzawi, M.; Crosby, A.; AlBalwi, M.A. Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am. J. Med. Genet. A 2013, 161A, 3155–3160. [Google Scholar] [CrossRef]
- Abu Asbeh, J.; Bystricka, A.; Qadir, M.; Nikolay, M.K.J. Raine Syndrome: Clinical and Radiological Features of a Case from the United Arab Emirates; Swiss Society of Neonatology: Switzerland, 2014. [Google Scholar]
- Seidahmed, M.Z.; Alazami, A.M.; Abdelbasit, O.B.; Al Hussein, K.; Miqdad, A.M.; Abu-Sa’da, O.; Mustafa, T.; Bahjat, S.; Alkuray, F.S. Report of a case of Raine syndrome and literature review. Am. J. Med. Genet. Part. A 2015, 167, 2394–2398. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.Y.; Rodriguez, M.; Roberts, A.; Bauer, M.; Mihalek, I.; Bodamer, O. A novel FAM20C mutation causes a rare form of neonatal lethal Raine syndrome. Am. J. Med. Genet. Part A 2019, 179, 1866–1871. [Google Scholar] [CrossRef] [PubMed]
- Wildeman, M.; Van Ophuizen, E.; Den Dunnen, J.T.; Taschner, P.E.M. Improving sequence variant descriptions in mutation databases and literature using the mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Silvertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 6220, 1260419. [Google Scholar] [CrossRef]
- Ishikawa, H.O.; Xu, A.; Ogura, E.; Manning, G.; Irvine, K.D. The raine syndrome protein FAM20C is a Golgi kinase that phosphorylates bio-mineralization proteins. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Dyle, M.C.; Kolakada, D.; Cortazar, M.A.; Jagannathan, S. How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated RNA decay. Wiley Interdiscip. Rev. RNA 2019. [Google Scholar] [CrossRef]
- Cui, J.; Xiao, J.; Tagliabracci, V.S.; Wen, J.; Rahdar, M.; Dixon, J.E. A secretory kinase complex regulates extracellular protein phosphorylation. Elife 2015, 4, e06120. [Google Scholar] [CrossRef] [Green Version]
- Fincham, A.G.; Moradian-Oldak, J.; Simmer, J.P. The structural biology of the developing dental enamel matrix. J. Struct. Biol. 1999, 126, 270–299. [Google Scholar] [CrossRef]
- Pollak, A.J.; Haghighi, K.; Kunduri, S.; Arvanitis, D.A.; Bidwell, P.A.; Liu, G.S.; Singh, V.P.; González, D.J.; Sanoudou, D.; Wiley, S.E.; et al. Phosphorylation of serine96 of histidine-rich calcium-binding protein by the Fam20C kinase functions to prevent cardiac arrhythmia. Proc. Natl. Acad. Sci. USA 2017, 114, 9098–9103. [Google Scholar] [CrossRef] [Green Version]
- Oya, K.; Ishida, K.; Nishida, T.; Sato, S.; Kishino, M.; Hirose, K.; Ogawa, Y.; Ikebe, K.; Takeshige, F.; Yasuda, H.; et al. Immunohistochemical analysis of dentin matrix protein 1 (Dmp1) phosphorylation by Fam20C in bone: Implications for the induction of biomineralization. Histochem. Cell Biol. 2017, 147, 341–351. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Hedge, A.M.; Rowe, P.S.N. Matrix extracellular phosphoglycoprotein (MEPE) is a new bone renal hormone and vascularization modulator. Endocrinology 2009, 150, 4012–4023. [Google Scholar] [CrossRef] [Green Version]
- Bergwitz, C.; Jüppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, S.; Li, C.; Gao, T.; Liu, Y.; Rangiani, A.; Sun, Y.; Hao, J.; George, A.; Lu, Y.; et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [Green Version]
- Andrukhova, O.; Bayer, J.; Schüler, C.; Zeitz, U.; Murali, S.K.; Ada, S.; Alvarez-Pez, J.M.; Smorodchenko, A.; Erben, R.G. Klotho lacks an FGF23-independent role in mineral homeostasis. J. Bone Miner. Res. 2017, 32, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Knab, V.M.; Corbin, B.; Andrukhova, O.; Hum, J.M.; Ni, P.; Rabadi, S.; Maeda, A.; Whyte, K.E.; Erben, R.G.; Jüppner, H.; et al. Acute Parathyroid hormone injection increases c-terminal but not intact fibroblast growth factor 23 levels. Endocrinology 2017, 158, 1130–1139. [Google Scholar] [CrossRef] [Green Version]
- Du, E.X.; Wang, X.F.; Yang, W.C.; Kaback, D.; Yee, S.P.; Qin, C.L.; George, A.; Hao, J.J. Characterization of Fam20C expression in odontogenesis and osteogenesis using transgenic mice. Int. J. Oral. Sci. 2015, 7, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jung, J.; Liu, Y.; Yuan, B.; Lu, Y.; Feng, J.Q.; Qin, C. The specific role of FAM20C in amelogenesis. J. Dent. Res. 2013, 92, 995–999. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RC | Year | Reference | Exon /Localization | c.description | p.description | M | Sp | I/NS | GR | KD |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2007 | Simpson et al. [11] | 7p22 | 45, XY psudic (7;7) (p22;p22) | - | X | X | |||
| 2 | 2007/ 1991 | Simpson et al. [11] /Kingston et al. [34] | E10 | c.1645C > T | p.Arg549Trp | X | X | |||
| 3 | 2007 | Simpson et al. [11] | E6 | c.1135G > A | p.Gly379Arg | X | X | |||
| 4 | 2007/ 2003 | Simpson et al. [11] Al-Gazali et al. [46] | I4–E5 | c.957-3C > G | Splicing | X | - | |||
| 5 | 2007/ 2003 | Simpson et al. [11] Hülskamp et al. [47] | E6 | c.1163T > G | p.Leu388Arg | X | X | |||
| 6 | 2007 | Simpson et al. [11] | E6/I7–E8 | c.1136G > A/c.1364-2A > G | p.Gly379Glu/Splicing | X | X | X | ||
| 7 | 2007 | Simpson et al. [11] | E4–I4/I8–E9 | c.956 + 5G > C/c.1446-1G > A | Splicing/Splicing | X/X | X/X | |||
| 8 | 2010 | Kochar et al. [48] | E10 | c.1672C > T | p.Arg558Trp | X | X | |||
| 9 | 2013 | Ababneh et al. [50] | 7p22 (48Kb) | 46,XY.ar[hg19] 7p22.3 (36480−523731)×0 | - | X | X | |||
| 10 | 2015 | Seidahmed et al. [52] | E6 | c.1225C > T | p.Arg409Cys | X | X | |||
| 11 | 2016 | Whyte et al. [44] | E6 | c.1094G > A | Gly365Asp | X | X | |||
| 12 | 2016 | Whyte et al. [44] | E6 | c.1094G > A | Gly365Asp | X | X | |||
| 13 | 2019 | Hung et al. [53] | E5 | c.1007T > G | p.Met336Arg | X | X |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Zavala, A.; Cortés-Camacho, F.; Palma-Lara, I.; Godínez-Aguilar, R.; Espinosa, A.M.; Pérez-Durán, J.; Villanueva-Ocampo, P.; Ugarte-Briones, C.; Serrano-Bello, C.A.; Sánchez-Santiago, P.J.; et al. Two Novel FAM20C Variants in a Family with Raine Syndrome. Genes 2020, 11, 222. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020222
Hernández-Zavala A, Cortés-Camacho F, Palma-Lara I, Godínez-Aguilar R, Espinosa AM, Pérez-Durán J, Villanueva-Ocampo P, Ugarte-Briones C, Serrano-Bello CA, Sánchez-Santiago PJ, et al. Two Novel FAM20C Variants in a Family with Raine Syndrome. Genes. 2020; 11(2):222. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020222
Chicago/Turabian StyleHernández-Zavala, Araceli, Fernando Cortés-Camacho, Icela Palma-Lara, Ricardo Godínez-Aguilar, Ana María Espinosa, Javier Pérez-Durán, Patricia Villanueva-Ocampo, Carlos Ugarte-Briones, Carlos Alberto Serrano-Bello, Paula Jesús Sánchez-Santiago, and et al. 2020. "Two Novel FAM20C Variants in a Family with Raine Syndrome" Genes 11, no. 2: 222. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020222