Hearing Impairment Overview in Africa: the Case of Cameroon

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection Criteria
2.2. Search Strategy
2.3. Selection of Studies
2.4. Data Extraction Process
2.5. Assessment of Methodological Quality
3. Results
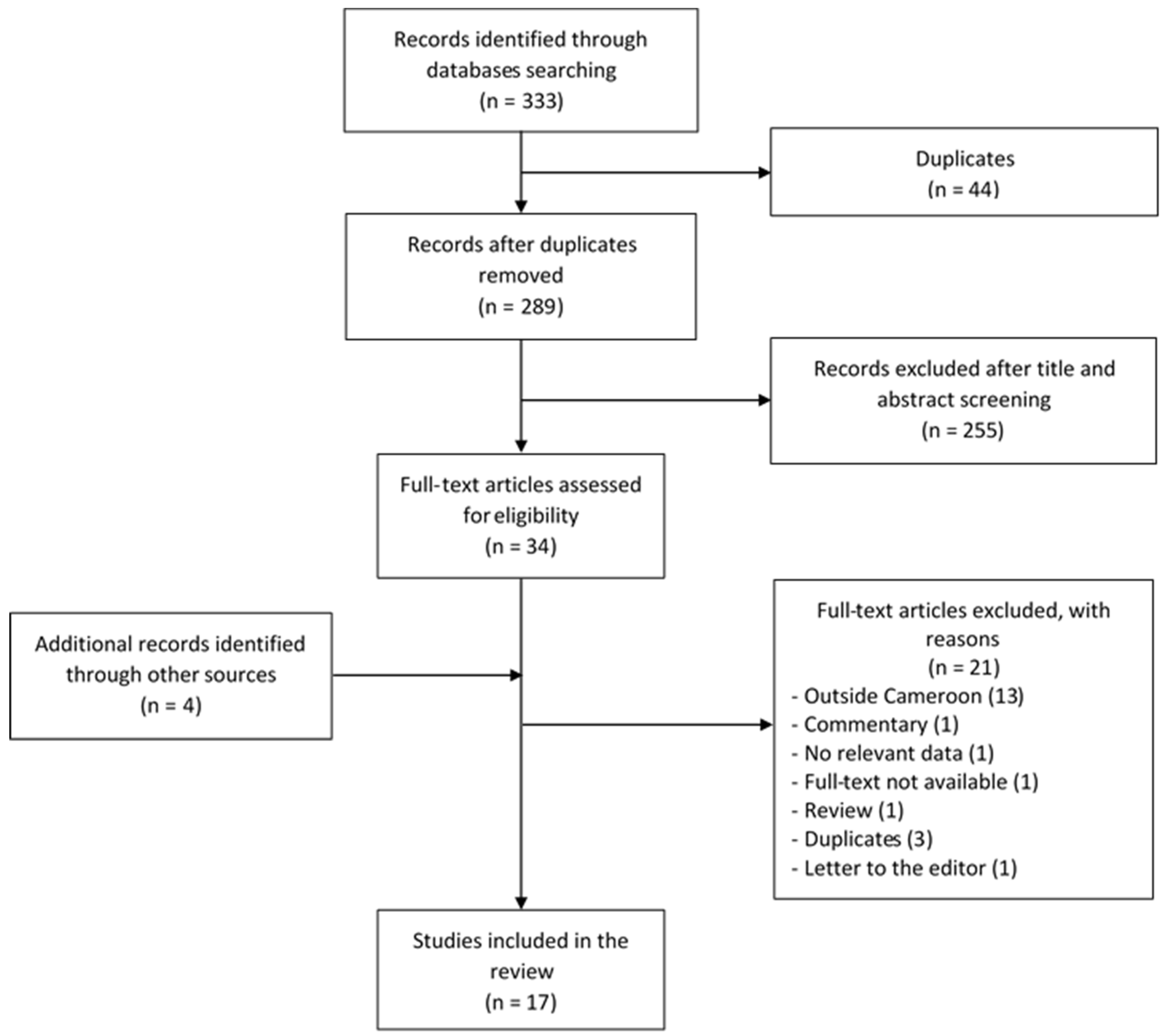
3.1. The Review Process
3.2. Characteristics of Included Studies
3.3. Prevalence of Hearing Impairment in Cameroon
3.4. Audiometric Characteristics of Hearing Impairment in Cameroon
3.5. Etiologies of Hearing Impairment in Cameroon
3.6. Hearing Impairment of Genetic Origin
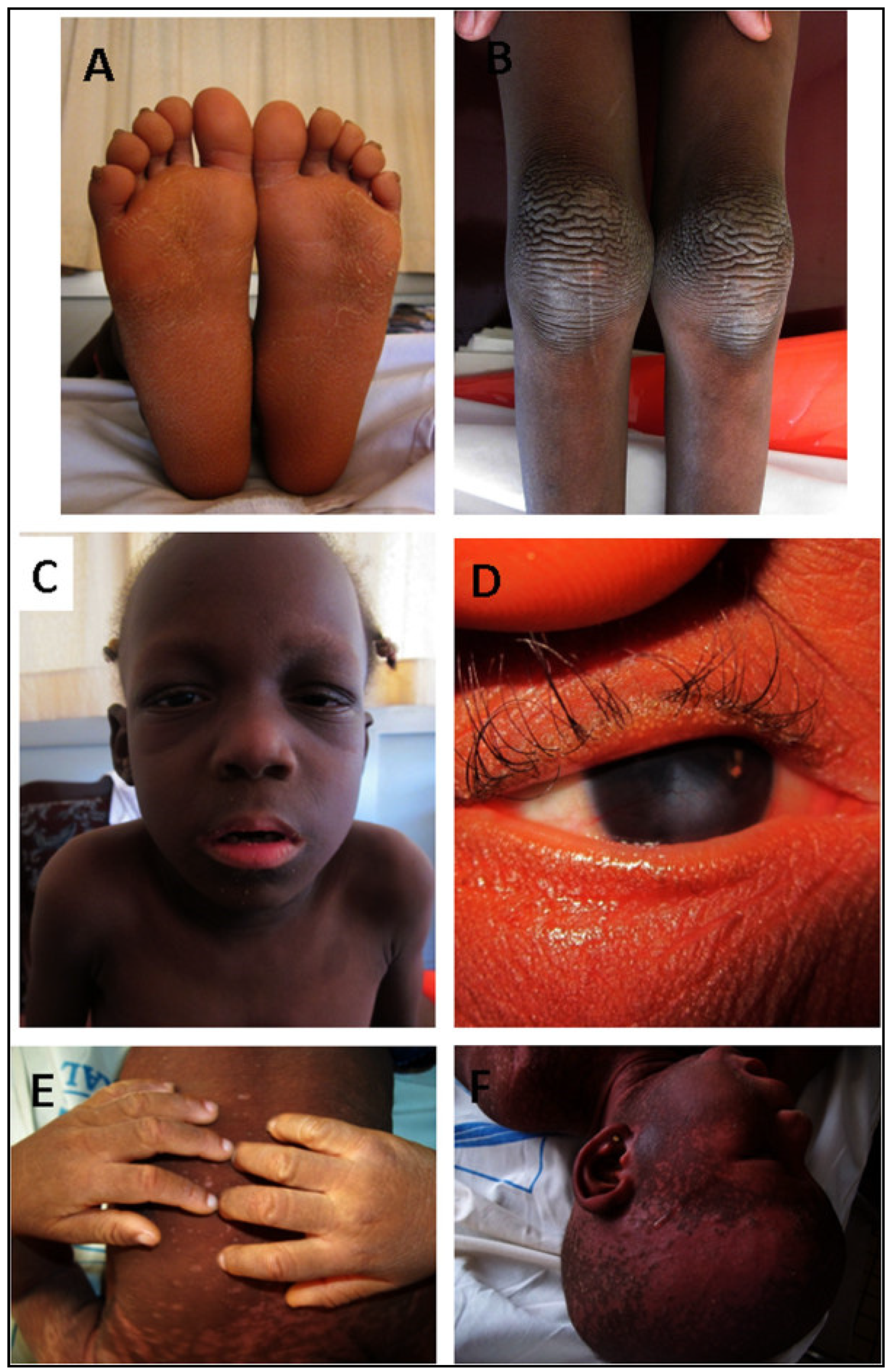
3.6.1. Clinical Patterns
3.6.2. Inheritance Pattern
3.6.3. Gene Variants and Hearing Impairment
Non-syndromic Hearing Impairment
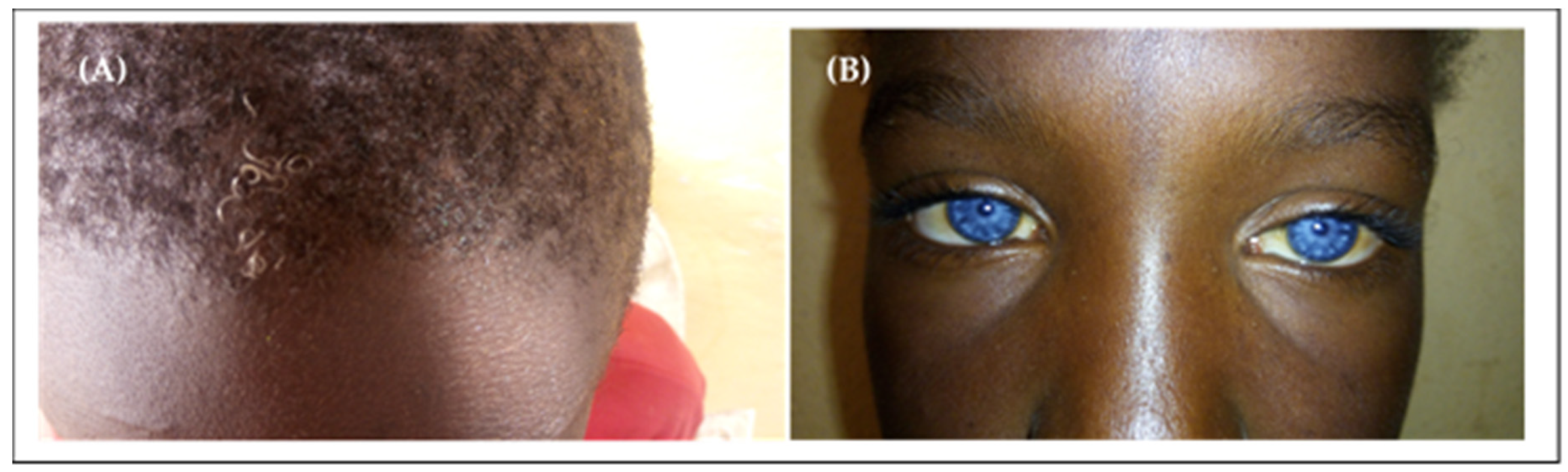
Keratitis–Ichtyosis–Deafness Syndrome
4. Discussion
5. Strengths and Limitations
6. Conclusions
7. Research Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Estimates. Available online: http://www.who.int/deafness/estimates/en/ (accessed on 29 December 2019).
- Mehra, S.; Eavey, R.D.; Keamy, D.G. The epidemiology of hearing impairment in the United States: Newborns, children, and adolescents. Otolaryngol. Head Neck Surg. 2009, 140, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Olusanya, B.O.; Neumann, K.J.; Saunders, J.E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 2014, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- James, M.; Kumar, P.; Ninan, P.J. A study on prevalence and risk factors of hearing impairment among newborns. Int. J. Contemporary Pediatr. 2018, 5, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Moctar, E.C.M.; Riahi, Z.; El Hachmi, H.; Veten, F.; Meiloud, G.; Bonnet, C.; Abdelhak, S.; Errami, M.; Houmeida, A. Etiology and associated GJB2 mutations in Mauritanian children with non-syndromic hearing loss. Eur. Arch. Otorhinolaryngol. 2016, 273, 3693–3698. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.D.O. The aetiology of childhood deafness in Sierra Leone. Sierra Leone Med. Dental Assoc. J. 1991, 6, 31–45. [Google Scholar]
- Lebeko, K.; Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Wonkam, A. Genetics of hearing loss in Africans: Use of next generation sequencing is the best way forward. Pan Afr. Med. J. 2015, 20, 383. [Google Scholar] [CrossRef]
- Grindle, C.R. Pediatric hearing loss. Pediatr. Rev. 2014, 35, 456–463. [Google Scholar] [CrossRef] [Green Version]
- Ječmenica, J.; Bajec-Opančina, A.; Ječmenica, D. Genetic hearing impairment. Childs Nerv. Syst. 2015, 31, 515–519. [Google Scholar] [CrossRef]
- Gürtler, N.; Lalwani, A.K. Etiology of syndromic and nonsyndromic sensorineural hearing loss. Otolaryngol. Clin. N. Am. 2002, 35, 891–908. [Google Scholar] [CrossRef]
- Bayazit, Y.A.; Yilmaz, M. An overview of hereditary hearing loss. ORL J. Otorhinolaryngol. Relat. Spec. 2006, 68, 57–63. [Google Scholar] [CrossRef]
- Hilgert, N.; Smith, R.J.H.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, P.-I.; Karltorp, E.; Carlsson-Hansén, E.; Åhlman, H.; Möller, C.; Vondöbeln, U. GJB2 (Connexin 26) gene mutations among hearing-impaired persons in a Swedish cohort. Acta Otolaryngol. 2012, 132, 1301–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansović, I.; Knezević, J.; Musani, V.; Seeman, P.; Barisić, I.; Pavelić, J. GJB2 mutations in patients with nonsyndromic hearing loss from Croatia. Genet. Test. Mol. Biomarkers 2009, 13, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Primignani, P.; Trotta, L.; Castorina, P.; Lalatta, F.; Sironi, F.; Radaelli, C.; Degiorgio, D.; Curcio, C.; Travi, M.; Ambrosetti, U.; et al. Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: Frequencies, novel mutations, genotypes, and degree of hearing loss. Genet. Test. Mol. Biomarkers 2009, 13, 209–217. [Google Scholar] [CrossRef]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Brobby, G.W.; Müller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W Mutation Associated with Recessive Nonsyndromic Sensorineural Deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef]
- Adadey, S.M.; Manyisa, N.; Mnika, K.; De Kock, C.; Nembaware, V.; Quaye, O.Q.; Amedofu, G.K.K.; Awandare, G.; Wonkam, A. GJB2 and GJB6 mutations in non-syndromic childhood hearing impairment in Ghana. Front. Genet. 2019, 10, 841. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020; 1999 February 14 [Updated 2017 July 27]. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1434/ (accessed on 14 February 2020).
- El-Serafy, G.M.; El-Bahnasawy, M.M.; Morsy, T.A. Management of hearing impairment in children. J. Egypt. Soc. Parasitol. 2013, 43, 333–340. [Google Scholar] [CrossRef]
- Stevens, G.; Flaxman, S.; Brunskill, E.; Mascarenhas, M.; Mathers, C.D.; Finucane, M. Global Burden of Disease Hearing Loss Expert Group Global and regional hearing impairment prevalence: An analysis of 42 studies in 29 countries. Eur. J. Public Health 2013, 23, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Mulwafu, W.; Kuper, H.; Ensink, R.J.H. Prevalence and causes of hearing impairment in Africa. Trop. Med. Int. Health 2016, 21, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohani, Z.N.; Meyre, D.; de Souza, R.J.; Joseph, P.G.; Gandhi, M.; Dennis, B.B.; Norman, G.; Anand, S.S. Assessing the quality of published genetic association studies in meta-analyses: The quality of genetic studies (Q-Genie) tool. BMC Genet. 2015, 16, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoy, D.; Brooks, P.; Woolf, A.; Blyth, F.; March, L.; Bain, C.; Baker, P.; Smith, E.; Buchbinder, R. Assessing risk of bias in prevalence studies: Modification of an existing tool and evidence of interrater agreement. J. Clin. Epidemiol. 2012, 65, 934–939. [Google Scholar] [CrossRef]
- Fokouo, J.V.F.; Vokwely, J.E.E.; Noubiap, J.J.N.; Nouthe, B.E.; Zafack, J.; Minka Ngom, E.S.; Dalil, A.B.; Ngo Nyeki, A.-R.; Bengono, G.; Njock, R. Effect of HIV Infection and Highly Active Antiretroviral Therapy on Hearing Function: A Prospective Case-Control Study from Cameroon. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wonkam, A.; Noubiap, J.J.N.; Djomou, F.; Fieggen, K.; Njock, R.; Toure, G.B. Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur. J. Med. Genet. 2013, 56, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Lebeko, K.; Manyisa, N.; Chimusa, E.R.; Mulder, N.; Dandara, C.; Wonkam, A. A Genomic and Protein-Protein Interaction Analyses of Nonsyndromic Hearing Impairment in Cameroon Using Targeted Genomic Enrichment and Massively Parallel Sequencing. OMICS 2017, 21, 90–99. [Google Scholar] [CrossRef]
- Tingang Wonkam, E.; Chimusa, E.; Noubiap, J.J.; Adadey, S.M.; F Fokouo, J.V.; Wonkam, A. GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon. Genes 2019, 10, 844. [Google Scholar] [CrossRef] [Green Version]
- Wonkam, A.; Noubiap, J.J.N.; Bosch, J.; Dandara, C.; Toure, G.B. Heterozygous p.Asp50Asn mutation in the GJB2 gene in two Cameroonian patients with keratitis-ichthyosis-deafness (KID) syndrome. BMC Med. Genet. 2013, 14, 81. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Lebeko, K.; Nziale, J.J.N.; Dandara, C.; Makubalo, N.; Wonkam, A. In search of genetic markers for nonsyndromic deafness in Africa: A study in Cameroonians and Black South Africans with the GJB6 and GJA1 candidate genes. OMICS 2014, 18, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Ferrite, S.; Mactaggart, I.; Kuper, H.; Oye, J.; Polack, S. Prevalence and causes of hearing impairment in Fundong Health District, North-West Cameroon. Trop. Med. Int. Health 2017, 22, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Makubalo, N.; Wright, G.; Entfellner, J.-B.D.; Tiffin, N.; Wonkam, A. Sequencing of GJB2 in Cameroonians and Black South Africans and comparison to 1000 Genomes Project Data Support Need to Revise Strategy for Discovery of Nonsyndromic Deafness Genes in Africans. OMICS 2014, 18, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeko, K.; Sloan-Heggen, C.M.; Noubiap, J.J.N.; Dandara, C.; Kolbe, D.L.; Ephraim, S.S.; Booth, K.T.; Azaiez, H.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 2016, 90, 288–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noubiap, J.-J.; Djomou, F.; Njock, R.; Toure, G.B.; Wonkam, A. Waardenburg syndrome in childhood deafness in Cameroon. S. Afr. J. Child Health 2014, 8, 3–5. [Google Scholar] [CrossRef]
- Trotta, L.; Iacona, E.; Primignani, P.; Castorina, P.; Radaelli, C.; Del Bo, L.; Coviello, D.; Ambrosetti, U. GJB2 and MTRNR1 contributions in children with hearing impairment from Northern Cameroon. Int. J. Audiol. 2011, 50, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Kuaban, C.; Noeske, J.; Rieder, H.L.; Aït-Khaled, N.; Abena Foe, J.L.; Trébucq, A. High effectiveness of a 12-month regimen for MDR-TB patients in Cameroon. Int. J. Tuberc. Lung Dis. 2015, 19, 517–524. [Google Scholar] [CrossRef]
- Trébucq, A.; Schwoebel, V.; Kashongwe, Z.; Bakayoko, A.; Kuaban, C.; Noeske, J.; Hassane, S.; Souleymane, B.; Piubello, A.; Ciza, F.; et al. Treatment outcome with a short multidrug-resistant tuberculosis regimen in nine African countries. Int. J. Tuberc. Lung Dis. 2018, 22, 17–25. [Google Scholar] [CrossRef]
- Djomou, F.; Nkouo, Y.C.A.; Mindja, E.D.; Nchinda, C.; Meka, L.; Mbamyah-Lyonga, E.; Ndjolo, A. Epidemiological and clinical aspects of ear nose and throat sensorineural emergencies in the Yaoundé reference hospital. Pan Afr. Med. J. 2016, 24, 251. [Google Scholar] [CrossRef]
- Jivraj, I.; Rudnisky, C.J.; Tambe, E.; Tipple, G.; Tennant, M.T.S. Identification of ocular and auditory manifestations of congenital rubella syndrome in Mbingo. Int. J. Telemed. Appl. 2014, 2014, 981312. [Google Scholar] [CrossRef] [Green Version]
- Chiabi, A.; Tchokoteu, P.F.; Toupouri, A.; Mbeng, T.B.; Wefuan, J. The clinical spectrum of severe malaria in children in the east provincial hospital of Bertoua, Cameroon. Bull. Soc. Pathol. Exot. 2004, 97, 239–243. [Google Scholar]
- Cockburn, L.; Cleaver, S.; Benuh, E. The Prevalence of Impairments and Disabilities in the North West Region, Cameroon. Health Sci. Dis. 2014, 15, 2. [Google Scholar]
- McPherson, B.; Holborow, C.A. A study of deafness in West Africa: The Gambian Hearing Health Project. Int. J. Pediatr. Otorhinolaryngol. 1985, 10, 115–135. [Google Scholar] [CrossRef]
- Ijaduola, G.T.A. The problems of the profoundly deaf Nigerian child. Postgrad. Dr.-Afr. 1982, 4, 180–184. [Google Scholar]
- Clark, J.L. Hearing loss in Mozambique: Current data from Inhambane Province. Int. J. Audiol. 2008, 47 (Suppl. 1), S49–S56. [Google Scholar] [CrossRef]
- Seely, D.R.; Gloyd, S.S.; Wright, A.D.; Norton, S.J. Hearing loss prevalence and risk factors among Sierra Leonean children. Arch. Otolaryngol. Head Neck Surg. 1995, 121, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, B.D.; Lee, P.K.; Lukwago, L.; Zaramba, S.; Bubikere, S.; Stewart, I. Cross-sectional survey of hearing impairment and ear disease in Uganda. J. Otolaryngol. Head Neck Surg. 2008, 37, 753–758. [Google Scholar] [PubMed]
- Louw, C.; Swanepoel, D.W.; Eikelboom, R.H.; Hugo, J. Prevalence of hearing loss at primary health care clinics in South Africa. Afr. Health Sci. 2018, 18, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Ramma, L.; Sebothoma, B. The prevalence of hearing impairment within the Cape Town Metropolitan area. S. Afr. J. Commun. Disord. 2016, 16, 105. [Google Scholar] [CrossRef]
- Fasunla, A.J.; Samdi, M.; Nwaorgu, O.G. An audit of Ear, Nose and Throat diseases in a tertiary health institution in South-western Nigeria. Pan Afr. Med. J. 2013, 14, 1. [Google Scholar] [CrossRef]
- Fischer, N.; Weber, B.; Riechelmann, H. [Presbycusis—Age Related Hearing Loss]. Laryngorhinootologie 2016, 95, 497–510. (In German) [Google Scholar]
- Gates, G.A.; Mills, J.H. Presbycusis. Lancet 2005, 366, 1111–1120. [Google Scholar] [CrossRef]
- Ologe, F.E.; Segun-Busari, S.; Abdulraheem, I.S.; Afolabi, A.O. Ear diseases in elderly hospital patients in Nigeria. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 404–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, G.R. Profound Childhood Deafness. J. Med. Genet. 1964, 1, 118–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellars, S.; Beighton, G.; Horan, F.; Beighton, P.H. Deafness in Black children is Southern Africa. S. Afr. Med. J. 1977, 51, 309–312. [Google Scholar] [PubMed]
- Hageman, M.J. Waardenburg’s syndrome in Kenyan Africans. Trop. Geogr. Med. 1978, 30, 45–55. [Google Scholar] [PubMed]
- de Saxe, M.; Kromberg, J.G.; Jenkins, T. Waardenburg syndrome in South Africa. Part I. An evaluation of the clinical findings in 11 families. S. Afr. Med. J. 1984, 66, 256–261. [Google Scholar] [PubMed]
- Otręba, M.; Miliński, M.; Buszman, E.; Wrześniok, D.; Beberok, A. [Hereditary hypomelanocytoses: The role of PAX3, SOX10, MITF, SNAI2, KIT, EDN3 and EDNRB genes]. Postepy Hig. Med. Dosw. (Online) 2013, 67, 1109–1118. (In Polish) [Google Scholar] [CrossRef]
- Read, A.P.; Newton, V.E. Waardenburg syndrome. J. Med. Genet. 1997, 34, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Bogdanova-Mihaylova, P.; Alexander, M.D.; Murphy, R.P.J.; Murphy, S.M. Waardenburg syndrome: A rare cause of inherited neuropathy due to SOX10 mutation. J. Peripher. Nerv. Syst. 2017, 22, 219–223. [Google Scholar] [CrossRef]
- Trabelsi, M.; Nouira, M.; Maazoul, F.; Kraoua, L.; Meddeb, R.; Ouertani, I.; Chelly, I.; Benoit, V.; Besbes, G.; Mrad, R. Novel PAX3 mutations causing Waardenburg syndrome type 1 in Tunisian patients. Int. J. Pediatr. Otorhinolaryngol. 2017, 103, 14–19. [Google Scholar] [CrossRef]
- Pampanos, A.; Economides, J.; Iliadou, V.; Neou, P.; Leotsakos, P.; Voyiatzis, N.; Eleftheriades, N.; Tsakanikos, M.; Antoniadi, T.; Hatzaki, A.; et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int. J. Pediatr. Otorhinolaryngol. 2002, 65, 101–108. [Google Scholar] [CrossRef]
- Dahl, H.H.; Saunders, K.; Kelly, T.M.; Osborn, A.H.; Wilcox, S.; Cone-Wesson, B.; Wunderlich, J.L.; Du Sart, D.; Kamarinos, M.; Gardner, R.J.; et al. Prevalence and nature of connexin 26 mutations in children with non-syndromic deafness. Med. J. Aust. 2001, 175, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Neocleous, V.; Aspris, A.; Shahpenterian, V.; Nicolaou, V.; Panagi, C.; Ioannou, I.; Kyamides, Y.; Anastasiadou, V.; Phylactou, L.A. High frequency of 35delG GJB2 mutation and absence of del (GJB6-D13S1830) in Greek Cypriot patients with nonsyndromic hearing loss. Genet. Test. 2006, 10, 285–289. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Cama, E.; Melchionda, S.; Palladino, T.; Carella, M.; Santarelli, R.; Genovese, E.; Benettazzo, F.; Zelante, L.; Arslan, E. Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int. J. Audiol. 2009, 48, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Bonyadi, M.J.; Fotouhi, N.; Esmaeili, M. Spectrum and frequency of GJB2 mutations causing deafness in the northwest of Iran. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Rudman, J.R.; Kabahuma, R.I.; Bressler, S.E.; Feng, Y.; Blanton, S.H.; Yan, D.; Liu, X.-Z. The genetic basis of deafness in populations of African descent. J. Genet. Genomics 2017, 44, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabahuma, R.I.; Ouyang, X.; Du, L.L.; Yan, D.; Hutchin, T.; Ramsay, M.; Penn, C.; Liu, X.-Z. Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Lasisi, A.O.; Bademci, G.; Foster, J.; Blanton, S.; Tekin, M. Common genes for non-syndromic deafness are uncommon in sub-Saharan Africa: A report from Nigeria. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1870–1873. [Google Scholar] [CrossRef] [Green Version]
- Gasmelseed, N.M.A.; Schmidt, M.; Magzoub, M.M.A.; Macharia, M.; Elmustafa, O.M.; Ototo, B.; Winkler, E.; Ruge, G.; Horstmann, R.D.; Meyer, C.G. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum. Mutat. 2004, 23, 206–207. [Google Scholar] [CrossRef]
- Javidnia, H.; Carson, N.; Awubwa, M.; Byaruhanga, R.; Mack, D.; Vaccani, J.-P. Connexin gene mutations among Ugandan patients with nonsyndromic sensorineural hearing loss. Laryngoscope 2014, 124, E373–E376. [Google Scholar] [CrossRef]
- Hu, S.; Sun, F.; Zhang, J.; Tang, Y.; Qiu, J.; Wang, Z.; Zhang, L. Genetic Etiology Study of Ten Chinese Families with Nonsyndromic Hearing Loss. Neural Plast. 2018, 2018, 4920980. [Google Scholar] [CrossRef] [PubMed]
- Mutai, H.; Suzuki, N.; Shimizu, A.; Torii, C.; Namba, K.; Morimoto, N.; Kudoh, J.; Kaga, K.; Kosaki, K.; Matsunaga, T. Diverse spectrum of rare deafness genes underlies early-childhood hearing loss in Japanese patients: A cross-sectional, multi-center next-generation sequencing study. Orphanet J. Rare Dis. 2013, 8, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Horta, O.; Duman, D.; Foster, J.; Sırmacı, A.; Gonzalez, M.; Mahdieh, N.; Fotouhi, N.; Bonyadi, M.; Cengiz, F.B.; Menendez, I.; et al. Whole-Exome Sequencing Efficiently Detects Rare Mutations in Autosomal Recessive Nonsyndromic Hearing Loss. PLoS ONE 2012, 7, e50628. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| First Author’s Name, Publication Year | Area | Regions | Study Setting | Study Design | Data Collection | Study Population | Male (%) | Mean Age (Years) | Age Range (Years) | Sample Size | Diagnosis Tool | Quality |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fokouo, 2015 [26] | urban | center region | hospital | case-control | prospective | patients followed up for HIV infection. | 28.3 | 33.4 ± 7.7 | 15–49 | 180 | PTA | high |
| Wonkam, 2013 [27] | urban and rural | 7 regions | school and hospital | cross sectional | prospective | patients with childhood deafness | 54.1 | 11 * | 1–32 | 582 | PTA and ABR | high |
| Lebeko, 2017 [28] | urban and rural | 7 regions | schools and hospital | cross sectional | prospective | patients with non-syndromic hearing impairment of either putative genetic origin or unknown origin | NR | NR | NR | 57 | PTA | high |
| Djomou, 2016 [39] | urban | center region | hospital | cross sectional | retrospective | patients admitted at the ENT unit for sensorineural emergencies | NR | 43.2 ± 17 | 16–66 | 22 | PTA | low |
| Tingang Wonkam, 2019 [29] | urban and rural | 8 regions | school and community | cross sectional | prospective | familial hearing impairment cases | 45.2 | 18 ± 10.4 | 1–50 | 93 | PTA and ABR | high |
| Trotta, 2011 [36] | rural | Far-North region | school | cross sectional | prospective | patients with prelingual hearing loss | 74.4 | NR | >5 | 70 | PTA | moderate |
| Wonkam, 2013 [30] | NR | NR | school | Case report | prospective | patients suffering from KID syndrome | 0 | 3.5 ± 2.12 | 2–5 | 2 | PTA and ABR | high |
| Kuaban, 2015 [37] | urban | 2 regions | hospital | longitudinal | prospective | MDR-TB (multidrug-resistant tuberculosis) patients, treated with a standardized 12-months regiment, including kanamycin. | 51.3 | 33.7 | 17–68 | 150 | PTA | moderate |
| Jivraj, 2014 [40] | rural | Northwest region | school | cross sectional | prospective | students at two schools, including a school for the deaf | 58.2 | 11.8 ± 2.8 | NR | 320 | NR | low |
| Bosch, 2014 [31] | urban and rural | 7 regions | school and hospital | cross sectional | prospective | patients with deafness of either putative genetic origin or unknown origin and that were shown not to have mutation in GJB2 gene | 52 | 12.11 | NR | 75 | PTA | high |
| Ferrite, 2017 [32] | rural | Northwest region | community | cross sectional | prospective | general population of the Fundong Health District, Northwest Cameroon | 40.8 | 24.4 | 0–80+ | 3567 | PTA and OAE | high |
| Bosch, 2014 [33] | urban and rural | 7 regions | school and hospital | cross sectional | prospective | patients with deafness of either putative genetic origin or unknown origin | NR | NR | NR | 180 | PTA | high |
| Lebeko, 2016 [34] | urban and rural | NR | school and hospital | cross sectional | prospective | families with at least two individuals with ARNSHI who were negative for pathogenic variants in GJB2 and GJB6 | 53.8 | NR | NR | 26 | PTA | high |
| Chiabi, 2004 [41] | rural | East region | hospital | cross sectional | retrospective | patients admitted and treated for severe malaria | 55.3 | 2.7 | 0–15 | 387 | self-reported | low |
| Trébucq, 2018 [38] | urban | NR | hospital | longitudinal | prospective | MDR-TB (multidrug-resistant tuberculosis) patients, treated with a standardized 9-months regiment, including kanamycin | NR | NR | ≥18 | 176 | PTA | moderate |
| Cockburn, 2014 [42] | rural | Northwest region | community | cross sectional | prospective | people living in the Northwest region of Cameroon | 43.3 | NR | 0–70+ | 18 878 | self-reported | low |
| Noubiap, 2014 [35] | urban and rural | 7 regions | schools and hospital | cross sectional | prospective | patients suffering from Waardenburg syndrome | 50 | 12.2 ± 7 | 6–25 | 6 | PTA | high |
| Country | Cameroon | Cameroon | Sierra Leone | Gambia | Ghana | Nigeria |
|---|---|---|---|---|---|---|
| Year of publication | 2013 | 2017 | 1991 | 1985 | 2019 | 1982 |
| Reference | [27] | [32] | [6] | [43] | [18] | [44] |
| Number of patients | 582 | 127 | 354 | 259 | 1104 | 298 |
| Hereditary | 14.8% | 0.8% | – | 8.1% | 21.3% | 13.1% |
| Meningitis | 34.4% | – | 23.9% | 31.7% | 3.9% | 11% |
| Impacted wax | – | 31.5% | – | – | – | – |
| Age-related HI | – | 22.8% | – | – | – | – |
| Noise-induced HI | – | 1.5% | – | – | – | – |
| Measles | 4.3% | – | 4.1% | 1.9% | 0.9% | 13% |
| Rubella | 0.5% | – | – | 1.5% | 0.2% | 2% |
| Mumps | 2.1% | – | 16.7% | – | 0.5% | 3% |
| Ototoxicity | 6% | – | 20.8% | – | – | 9% |
| Other | 5.3% | 6.4% | – | 2.3 | 13.1% | 7.7% |
| Unknown | 32.6% | 37% | 34.8% | 54.4% | 60.1% | 41.2% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wonkam Tingang, E.; Noubiap, J.J.; F. Fokouo, J.V.; Oluwole, O.G.; Nguefack, S.; Chimusa, E.R.; Wonkam, A. Hearing Impairment Overview in Africa: the Case of Cameroon. Genes 2020, 11, 233. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020233
Wonkam Tingang E, Noubiap JJ, F. Fokouo JV, Oluwole OG, Nguefack S, Chimusa ER, Wonkam A. Hearing Impairment Overview in Africa: the Case of Cameroon. Genes. 2020; 11(2):233. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020233
Chicago/Turabian StyleWonkam Tingang, Edmond, Jean Jacques Noubiap, Jean Valentin F. Fokouo, Oluwafemi Gabriel Oluwole, Séraphin Nguefack, Emile R. Chimusa, and Ambroise Wonkam. 2020. "Hearing Impairment Overview in Africa: the Case of Cameroon" Genes 11, no. 2: 233. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020233