Reversion to Normal of FMR1 Expanded Alleles: A Rare Event in Two Independent Fragile X Syndrome Families

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. CGG Sizing and Methylation Analysis
2.3. Haplotype Analysis
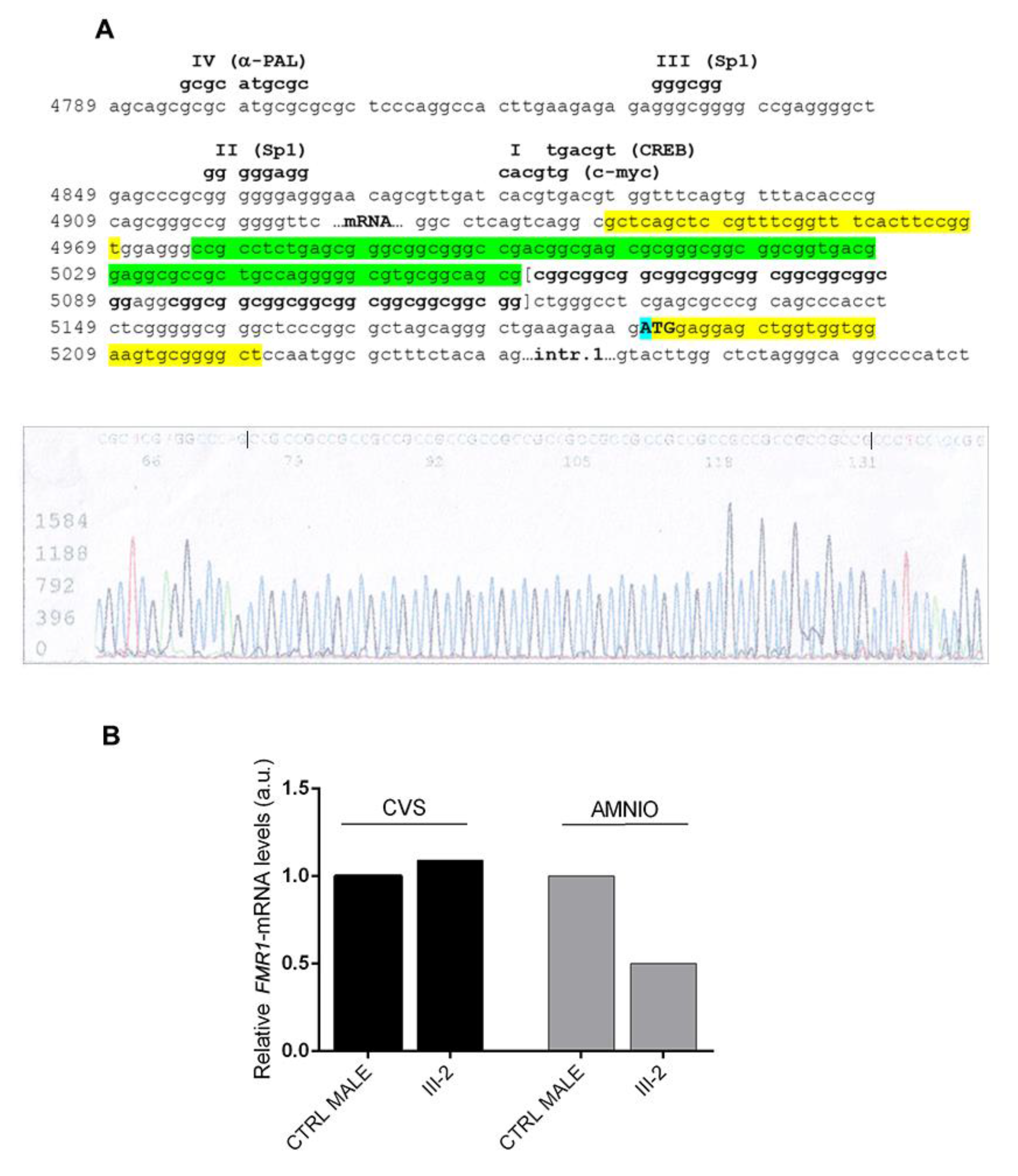
2.4. Sequencing Analysis
2.5. FMR1-mRNA Quantification
3. Results
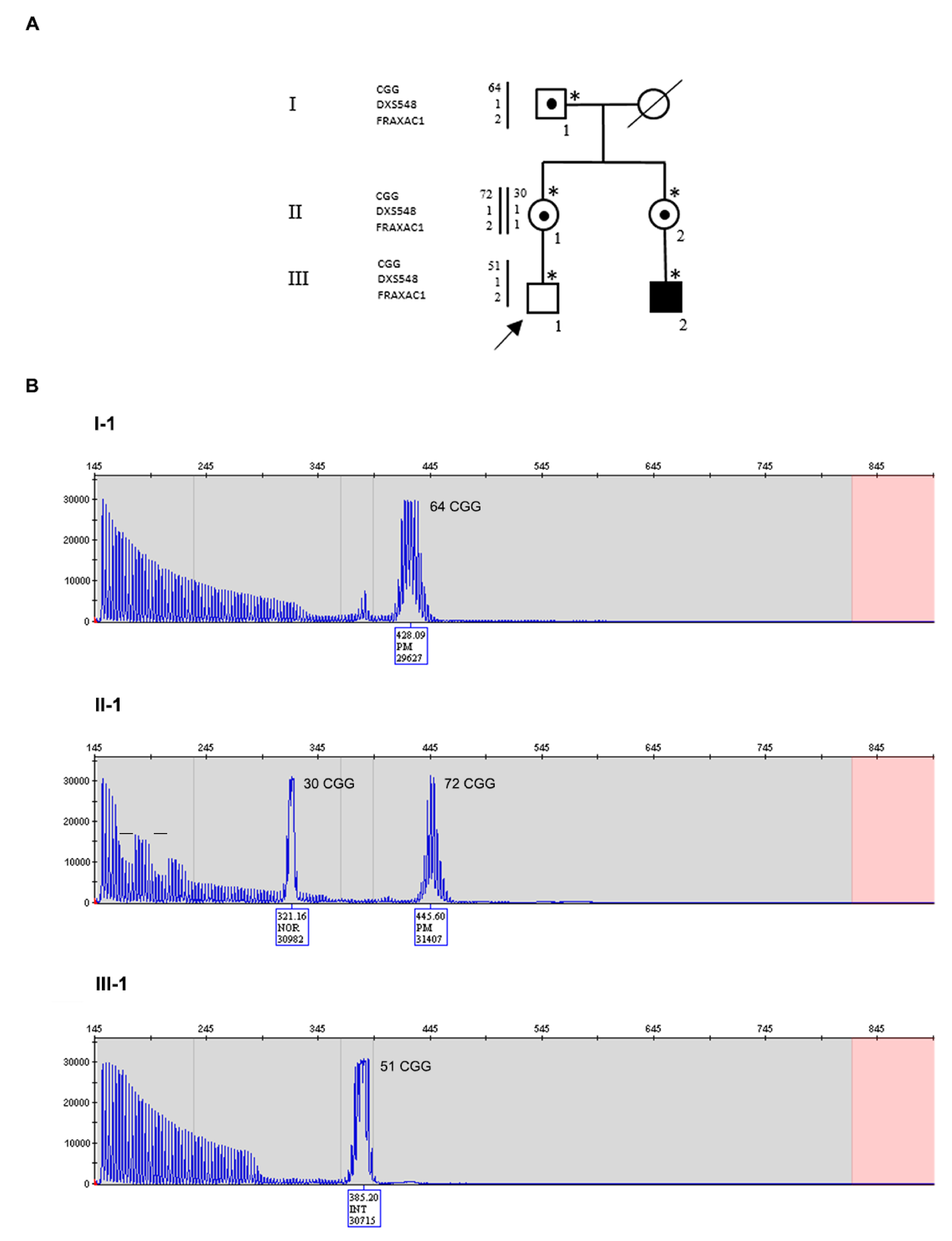
3.1. Family 1
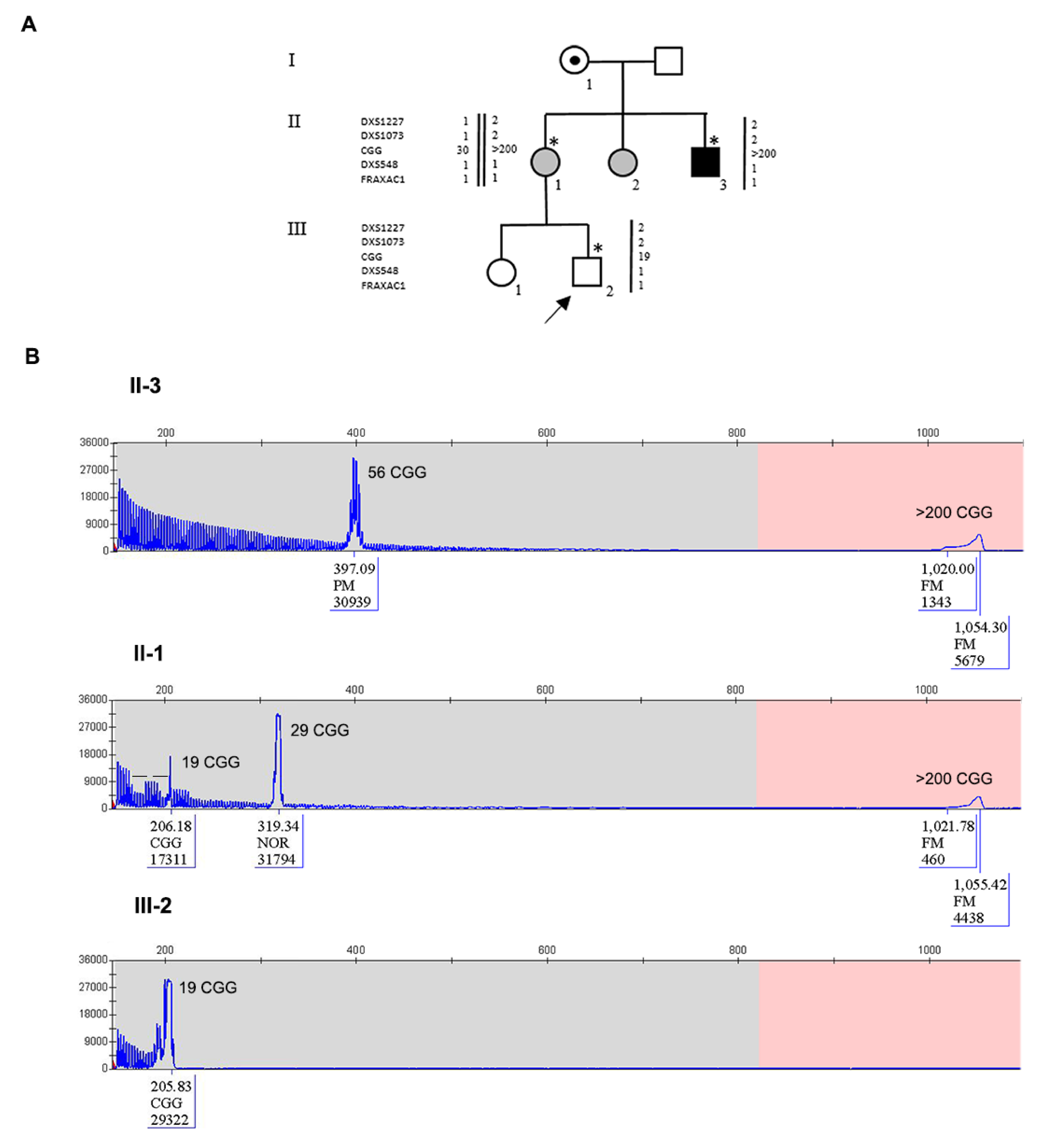
3.2. Family 2
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pirozzi, F.; Tabolacci, E.; Neri, G. The FRAXopathies: Definition, overview, and update. Am. J. Med. Genet. A 2011, 155, 1803–1816. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef]
- Allingham-Hawkins, D.J.; Babul-Hirji, R.; Chitayat, D.; Holden, J.J.; Yang, K.T.; Lee, C.; Hudson, R.; Gorwill, H.; Nolin, S.L.; Glicksman, A.; et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The international collaborative pof in fragile x study—Preliminary data. Am. J. Med. Genet. 1999, 83, 322–325. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Tassone, F.; Iong, K.P.; Tong, T.H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2013, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Bontekoe, C.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; van der Linde, H.; Lans, H.; de Lange, D.; Hirst, M.C.; Oostra, B.A. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum. Mol. Genet. 2001, 10, 1693–1699. [Google Scholar] [CrossRef] [Green Version]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef]
- Sullivan, A.K.; Crawford, D.C.; Scott, E.H.; Leslie, M.L.; Sherman, S.L. Paternally transmitted FMR1 alleles are less stable than maternally transmitted alleles in the common and intermediate size range. Am. J. Hum. Genet. 2002, 70, 1532–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberlé, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Genuardi, M.; Kozak, L.; Giovannucci-Uzielli, M.L.; Bussani, C.; Dagna-Bricarelli, F.; Grasso, M.; Perroni, L.; Sebastio, G.; Sperandeo, M.P.; et al. Fragile X founder chromosomes in Italy: A few initial events and possible explanation for their heterogeneity. Am. J. Med. Genet. 1996, 64, 209–215. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. Ups and Downs: Mechanisms of Repeat Instability in the Fragile X-Related Disorders. Genes (Basel) 2016, 7, 70. [Google Scholar] [CrossRef]
- Reyniers, E.; Vits, L.; De Boulle, K.; Van Roy, B.; Van Velzen, D.; de Graaff, E.; Verkerk, A.J.; Jorens, H.Z.; Darby, J.K.; Oostra, B.; et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 1993, 4, 143–146. [Google Scholar] [CrossRef]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J.; et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Alfaro, M.P.; Cohen, M.; Vnencak-Jones, C.L. Maternal FMR1 premutation allele expansion and contraction in fraternal twins. Am. J. Med. Genet. A 2013, 161, 2620–2625. [Google Scholar]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 2019, 179, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Tabolacci, E.; Pomponi, M.G.; Pietrobono, R.; Chiurazzi, P.; Neri, G. A unique case of reversion to normal size of a maternal premutation FMR1 allele in a normal boy. Eur. J. Hum. Genet. 2008, 16, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, M.; Grasbon-Frodl, E.; Neitzel, B.; Kooy, F.; Holinski-Feder, E. FMR1 gene deletion/reversion: A pitfall of fragile X carrier testing. Genet. Test 2003, 7, 303–308. [Google Scholar] [CrossRef]
- Manor, E.; Jabareen, A.; Magal, N.; Kofman, A.; Hagerman, R.J.; Tassone, F. Prenatal Diagnosis of Fragile X: Can a Full Mutation Allele in the FMR1 Gene Contract to a Normal Size? Front. Genet. 2017, 8, 158. [Google Scholar] [CrossRef] [Green Version]
- Prawer, Y.; Hunter, M.; Cronin, S.; Ling, L.; Aliaga Vera, S.; Fahey, M.; Gelfand, N.; Oertel, R.; Bartlett, E.; Francis, D.; et al. Prenatal diagnosis of fragile x syndrome in a twin pregnancy complicated by a complete retraction. Genes (Basel) 2018, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Maia, N.; Loureiro, J.R.; Oliveira, B.; Marques, I.; Santos, R.; Jorge, P.; Martins, S. Contraction of fully expanded FMR1 alleles to the normal range: Predisposing haplotype or rare events? J. Hum. Genet. 2017, 62, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Pandelache, A.; Baker, E.K.; Aliaga, S.M.; Arpone, M.; Forbes, R.; Stark, Z.; Francis, D.; Godler, D.E. Clinical and Molecular Differences between 4-Year-Old Monozygous Male Twins Mosaic for Normal, Premutation and Fragile X Full Mutation Alleles. Genes (Basel) 2019, 10, 279. [Google Scholar] [CrossRef] [Green Version]
- Kambouris, M.; Snow, K.; Thibodeau, S.; Bluhm, D.; Green, M.; Feldman, G.L. Segregation of the fragile X mutation from a male with a full mutation: Unusual somatic instability in the FMR-1 locus. Am. J. Med. Genet. 1996, 64, 404–407. [Google Scholar] [CrossRef]
- Dobkin, C.S.; Nolin, S.L.; Cohen, I.; Sudhalter, V.; Bialer, M.G.; Ding, X.H.; Jenkins, E.C.; Zhong, N.; Brown, W.T. Tissue differences in fragile X mosaics: Mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 1996, 64, 296–301. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef]
- Grønskov, K.; Hjalgrim, H.; Bjerager, M.O.; Brøndum-Nielsen, K. Deletion of all CGG repeats plus flanking sequences in FMR1 does not abolish gene expression. Am. J. Hum. Genet. 1997, 61, 961–967. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabolacci, E.; Pietrobono, R.; Maneri, G.; Remondini, L.; Nobile, V.; Della Monica, M.; Pomponi, M.G.; Genuardi, M.; Neri, G.; Chiurazzi, P. Reversion to Normal of FMR1 Expanded Alleles: A Rare Event in Two Independent Fragile X Syndrome Families. Genes 2020, 11, 248. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030248
Tabolacci E, Pietrobono R, Maneri G, Remondini L, Nobile V, Della Monica M, Pomponi MG, Genuardi M, Neri G, Chiurazzi P. Reversion to Normal of FMR1 Expanded Alleles: A Rare Event in Two Independent Fragile X Syndrome Families. Genes. 2020; 11(3):248. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030248
Chicago/Turabian StyleTabolacci, Elisabetta, Roberta Pietrobono, Giulia Maneri, Laura Remondini, Veronica Nobile, Matteo Della Monica, Maria Grazia Pomponi, Maurizio Genuardi, Giovanni Neri, and Pietro Chiurazzi. 2020. "Reversion to Normal of FMR1 Expanded Alleles: A Rare Event in Two Independent Fragile X Syndrome Families" Genes 11, no. 3: 248. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030248