Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Cell line Growth and DNA Extraction
2.2. Determination of Chimpanzee Subspecies
2.3. ONT Promethion Library Preparation and Sequencing
2.4. BNG Saphyr Library Preparation and Sequencing
2.5. Detection of SVs
2.6. Genotyping and Filtering of SVs
2.7. Annotation of Impacted Genes
2.8. Differential Gene Expression
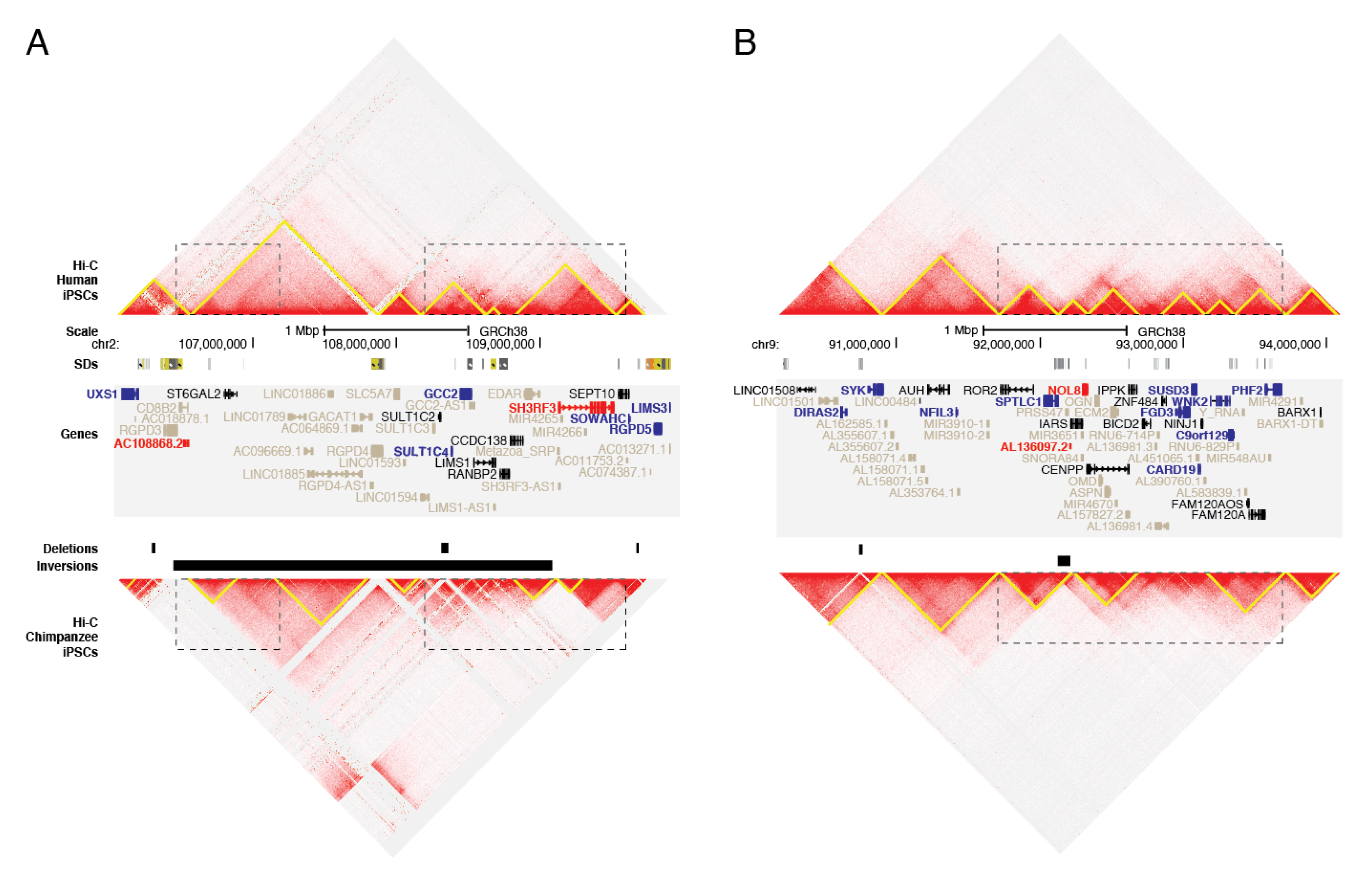
2.9. Topologically-Associated Domain (TAD) Analyses
2.10. Permutation Analyses
3. Results
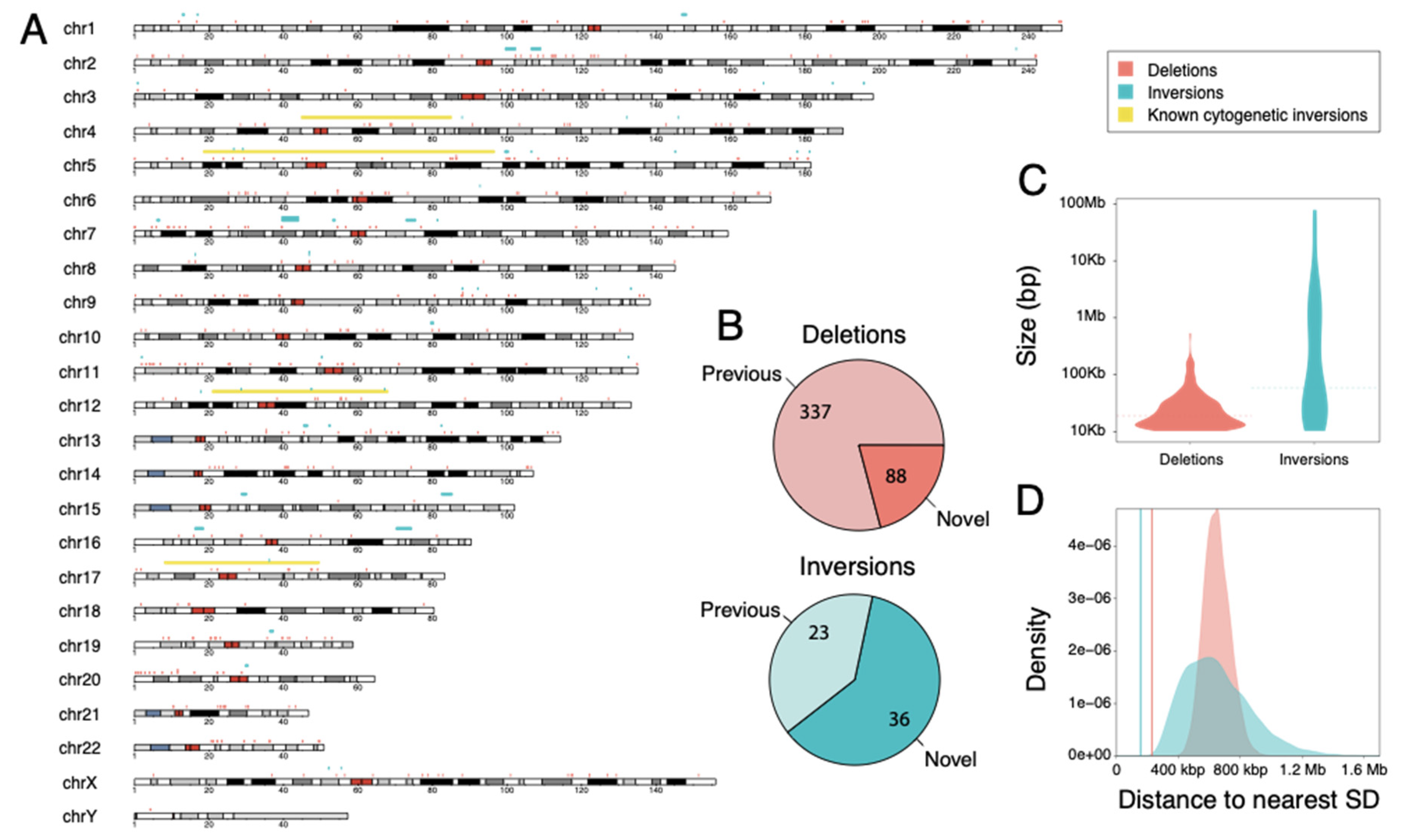
3.1. Large-Scale SV Discovery and Genotyping in Chimpanzee
3.2. Genomic Features of Identified SVs
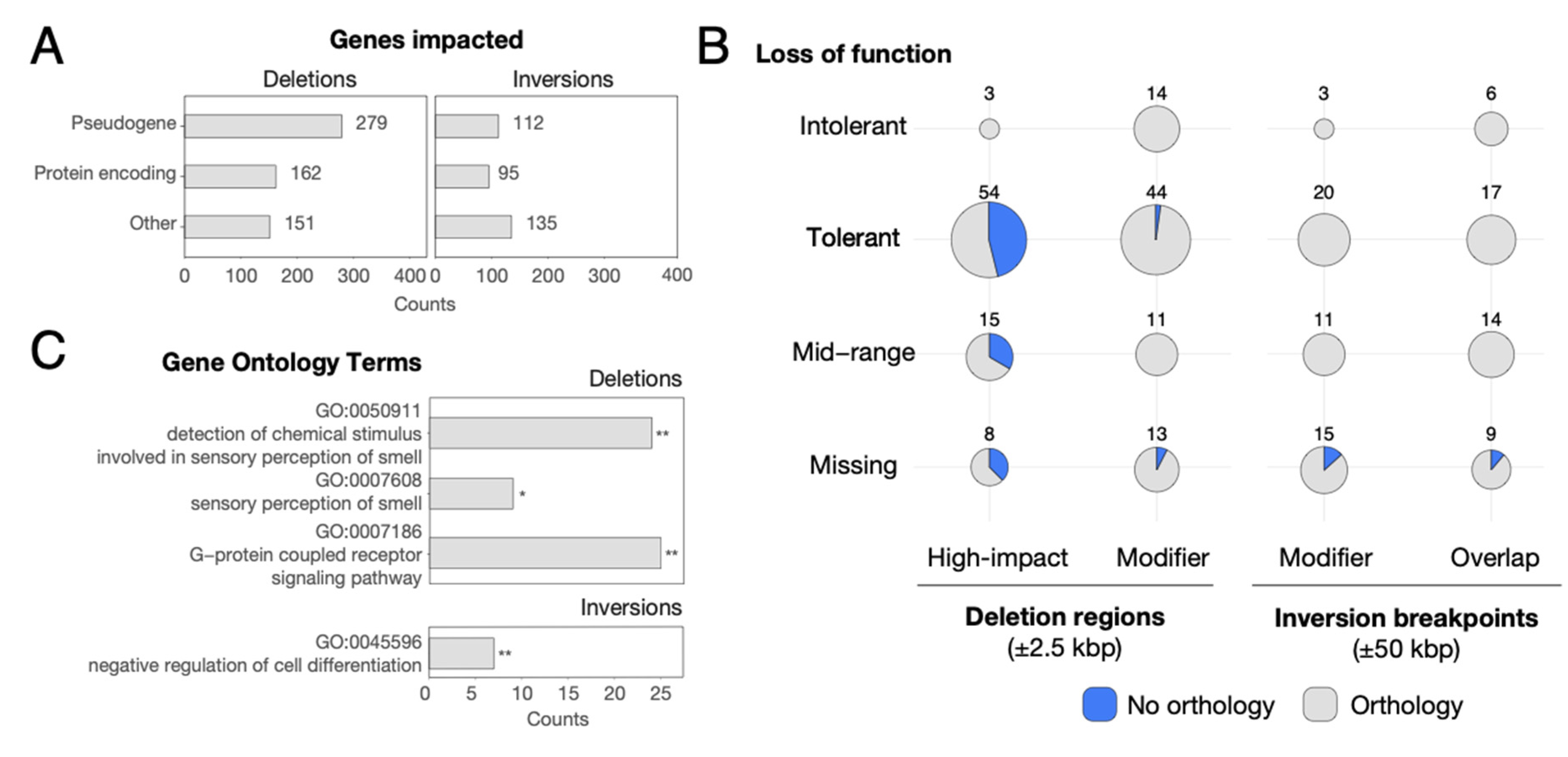
3.3. Genes Impacted by SVs
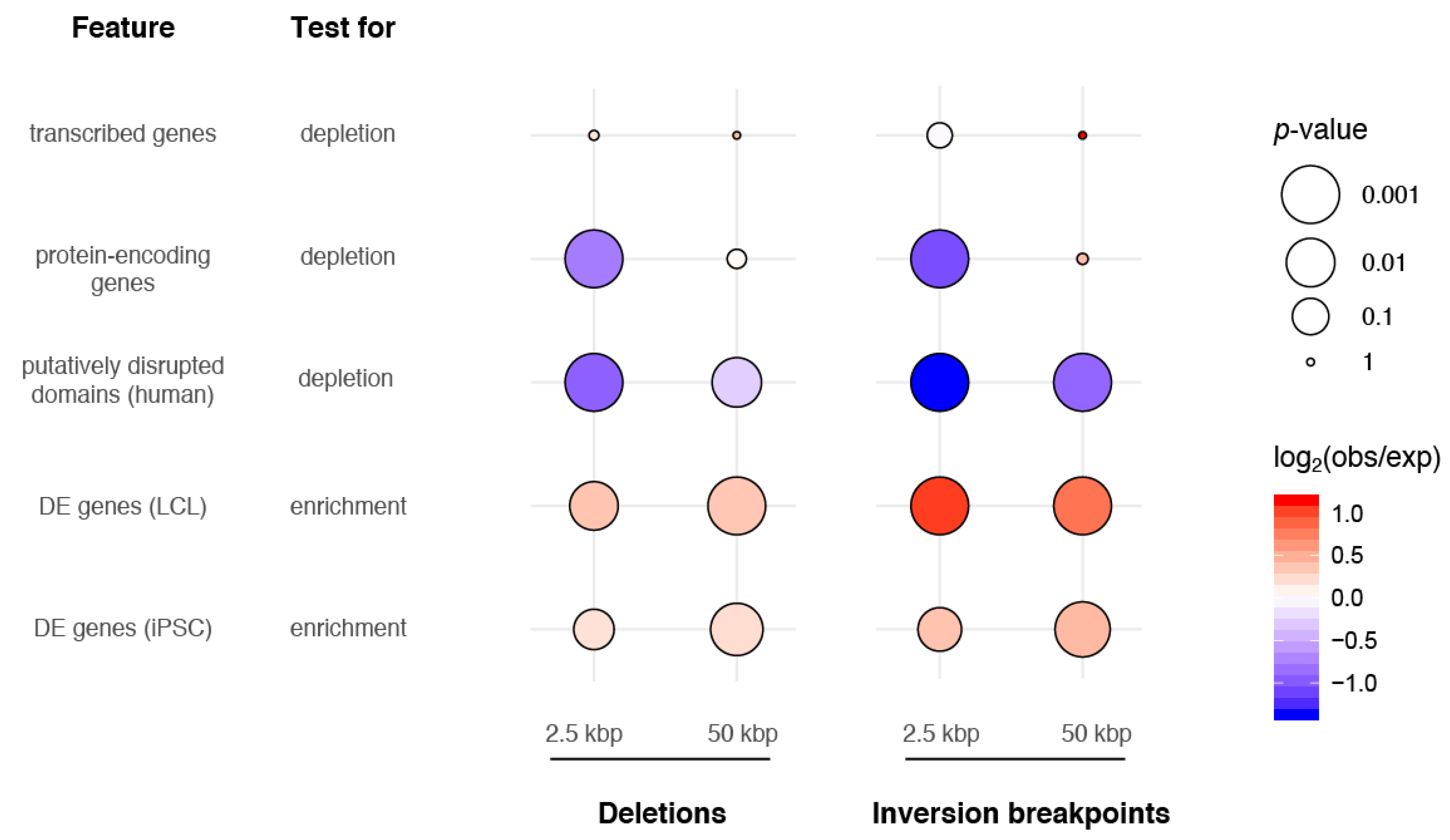
3.4. SVs and Gene Regulation
3.5. Genes Showing Signatures of Natural Selection
3.6. Genes Impacted by Chimpanzee-Specific SVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Patterson, N.; Richter, D.J.; Gnerre, S.; Lander, E.S.; Reich, D. Genetic evidence for complex speciation of humans and chimpanzees. Nature 2006, 441, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Langergraber, K.E.; Prüfer, K.; Rowney, C.; Boesch, C.; Crockford, C.; Fawcett, K.; Inoue, E.; Inoue-Muruyama, M.; Mitani, J.C.; Muller, M.N.; et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15716–15721. [Google Scholar] [CrossRef] [Green Version]
- O’Bleness, M.; Searles, V.B.; Varki, A.; Gagneux, P.; Sikela, J.M. Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet. 2012, 13, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A. Comparing the human and chimpanzee genomes: Searching for needles in a haystack. Genome Res. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado-Martinez, J.; Sudmant, P.H.; Kidd, J.M.; Li, H.; Kelley, J.L.; Lorente-Galdos, B.; Veeramah, K.R.; Woerner, A.E.; O’Connor, T.D.; Santpere, G.; et al. Great ape genetic diversity and population history. Nature 2013, 499, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Gallego Romero, I.; Pavlovic, B.J.; Hernando-Herraez, I.; Zhou, X.; Ward, M.C.; Banovich, N.E.; Kagan, C.L.; Burnett, J.E.; Huang, C.H.; Mitrano, A.; et al. A panel of induced pluripotent stem cells from chimpanzees: A resource for comparative functional genomics. eLife 2015, 4, e07103. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.; Ford, M.J.; Cusanovich, D.A.; Mitrano, A.; Pritchard, J.K.; Gilad, Y. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 2013, 342, 1100–1104. [Google Scholar] [CrossRef] [Green Version]
- McLean, C.Y.; Reno, P.L.; Pollen, A.A.; Bassan, A.I.; Capellini, T.D.; Guenther, C.; Indjeian, V.B.; Lim, X.; Menke, D.B.; Schaar, B.T.; et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 2011, 471, 216–219. [Google Scholar] [CrossRef]
- Prescott, S.L.; Srinivasan, R.; Marchetto, M.C.; Grishina, I.; Narvaiza, I.; Selleri, L.; Gage, F.H.; Swigut, T.; Wysocka, J. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 2015, 163, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Pollen, A.A.; Bhaduri, A.; Andrews, M.G.; Nowakowski, T.J.; Meyerson, O.S.; Mostajo-Radji, M.A.; Di Lullo, E.; Alvarado, B.; Bedolli, M.; Dougherty, M.L.; et al. Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 2019, 176, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csardi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef]
- Eres, I.E.; Luo, K.; Hsiao, C.J.; Blake, L.E.; Gilad, Y. Reorganization of 3D genome structure may contribute to gene regulatory evolution in primates. PLoS Genet. 2019, 15, e1008278. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Cain, C.E.; Myrthil, M.; Lewellen, N.; Michelini, K.; Davenport, E.R.; Stephens, M.; Pritchard, J.K.; Gilad, Y. Epigenetic modifications are associated with inter-species gene expression variation in primates. Genome Biol. 2014, 15, 547. [Google Scholar] [CrossRef]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar] [CrossRef]
- Rogers, J.; Gibbs, R.A. Comparative primate genomics: Emerging patterns of genome content and dynamics. Nat. Rev. Genet. 2014, 15, 347–359. [Google Scholar] [CrossRef]
- Newman, T.L.; Tuzun, E.; Morrison, V.A.; Hayden, K.E.; Ventura, M.; McGrath, S.D.; Rocchi, M.; Eichler, E.E. A genome-wide survey of structural variation between human and chimpanzee. Genome Res. 2005, 15, 1344–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokcumen, O.; Tischler, V.; Tica, J.; Zhu, Q.; Iskow, R.C.; Lee, E.; Fritz, M.H.-Y.; Langdon, A.; Stütz, A.M.; Pavlidis, P.; et al. Primate genome architecture influences structural variation mechanisms and functional consequences. Proc. Natl. Acad. Sci. USA 2013, 110, 15764–15769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.M.; Flibotte, S.; Missirlis, P.I.; Marra, M.A.; Jones, S.; Thornton, K.; Clark, A.G.; Holt, R.A. Identification by full-coverage array CGH of human DNA copy number increases relative to chimpanzee and gorilla. Genome Res. 2006, 16, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Locke, D.P.; Segraves, R.; Carbone, L.; Archidiacono, N.; Albertson, D.G.; Pinkel, D.; Eichler, E.E. Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome Res. 2003, 13, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskow, R.C.; Gokcumen, O.; Abyzov, A.; Malukiewicz, J.; Zhu, Q.; Ann, T.; Athma, S.; Pai, A. Regulatory Element Copy Number Differences Shape Primate Expression Profiles. Proc. Natl. Acad. Sci. USA 2012, 109, 12656–12661. [Google Scholar] [CrossRef] [Green Version]
- Marques-Bonet, T.; Kidd, J.M.; Ventura, M.; Graves, T.A.; Cheng, Z.; Hillier, L.W.; Jiang, Z.; Baker, C.; Malfavon-Borja, R.; Fulton, L.A.; et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature 2009, 457, 877–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Huddleston, J.; Catacchio, C.R.; Malig, M.; Hillier, L.W.; Baker, C.; Mohajeri, K.; Kondova, I.; Bontrop, R.E.; Persengiev, S.; et al. Evolution and diversity of copy number variation in the great ape lineage. Genome Res. 2013, 23, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catacchio, C.R.; Maggiolini, F.A.M.; D’Addabbo, P.; Bitonto, M.; Capozzi, O.; Lepore Signorile, M.; Miroballo, M.; Archidiacono, N.; Eichler, E.E.; Ventura, M.; et al. Inversion variants in human and primate genomes. Genome Res. 2018, 28, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuk, L.; MacDonald, J.R.; Tang, T.; Carson, A.R.; Li, M.; Rao, G.; Khaja, R.; Scherer, S.W. Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet. 2005, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Kuderna, L.F.K.; Tomlinson, C.; Hillier, L.W.; Tran, A.; Fiddes, I.T.; Armstrong, J.; Laayouni, H.; Gordon, D.; Huddleston, J.; Garcia Perez, R.; et al. A 3-way hybrid approach to generate a new high-quality chimpanzee reference genome (Pan_tro_3.0). Gigascience 2017, 6, gix098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronenberg, Z.N.; Fiddes, I.T.; Gordon, D.; Murali, S.; Cantsilieris, S.; Meyerson, O.S.; Underwood, J.G.; Nelson, B.J.; Chaisson, M.J.P.; Dougherty, M.L.; et al. High-resolution comparative analysis of great ape genomes. Science 2018, 360. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019. [Google Scholar] [CrossRef]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [Green Version]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Manuel, M.; Kuhlwilm, M.; Frandsen, P.; Sousa, V.C.; Desai, T.; Prado-Martinez, J.; Hernandez-Rodriguez, J.; Dupanloup, I.; Lao, O.; Hallast, P.; et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 2016, 354, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, A.R.; Lam, E.T.; Pang, A.W.C.; Zhang, X.; Andrews, W.; Lee, J.; Liang, T.Y.; Wang, J.; Zhou, X.; Zhu, Z.; et al. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Next-Generation Mapping. BioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2015. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Cagan, A.; Theunert, C.; Laayouni, H.; Santpere, G.; Pybus, M.; Casals, F.; Prüfer, K.; Navarro, A.; Marques-Bonet, T.; Bertranpetit, J.; et al. Natural Selection in the Great Apes. Mol. Biol. Evol. 2016, 33, 3268–3283. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, B.J.; Blake, L.E.; Roux, J.; Chavarria, C.; Gilad, Y. A Comparative Assessment of Human and Chimpanzee iPSC-derived Cardiomyocytes with Primary Heart Tissues. Sci. Rep. 2018, 8, 15312. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Michael, I.; Rafael, L.; Irizarry, A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Charity, W.; Shi, L.W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, V.; Cusanovich, D.A.; Hause, R.J.; Ma, W.; Qiu, R.; Deng, X.; Blau, C.A.; Disteche, C.M.; Noble, W.S.; Shendure, J.; et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nat. Protoc. 2016, 11, 2104–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Shi, Y.; Dai, C.; Tjong, H.; Gong, K.; Alber, F.; Zhou, X.J. TopDom: An efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016, 44, e70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, M.; Tavernari, D.; Oricchio, E.; Ciriello, G. Comparison of computational methods for the identification of topologically associating domains. Genome Biol. 2018, 19, 217. [Google Scholar] [CrossRef] [Green Version]
- McVicker, G.; van de Geijn, B.; Degner, J.F.; Cain, C.E.; Banovich, N.E.; Raj, A.; Lewellen, N.; Myrthil, M.; Gilad, Y.; Pritchard, J.K. Identification of genetic variants that affect histone modifications in human cells. Science 2013, 342, 747–749. [Google Scholar] [CrossRef] [Green Version]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome Structural Variation Discovery and Genotyping. Nat. Rev. Genet. 2011. [Google Scholar] [CrossRef]
- Nickerson, E.; Nelson, D.L. Molecular definition of pericentric inversion breakpoints occurring during the evolution of humans and chimpanzees. Genomics 1998, 50, 368–372. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Sandig, C.; Chuzhanova, N.; Goidts, V.; Szamalek, J.M.; Tänzer, S.; Müller, S.; Platzer, M.; Cooper, D.N.; Hameister, H. Breakpoint analysis of the pericentric inversion distinguishing human chromosome 4 from the homologous chromosome in the chimpanzee (Pan troglodytes). Hum. Mutat. 2005, 25, 45–55. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Sandig, C.A.; Goidts, V.; Hameister, H. Breakpoint analysis of the pericentric inversion between chimpanzee chromosome 10 and the homologous chromosome 12 in humans. Cytogenet. Genome Res. 2005, 108, 91–97. [Google Scholar] [CrossRef]
- Goidts, V.; Szamalek, J.M.; de Jong, P.J.; Cooper, D.N.; Chuzhanova, N.; Hameister, H.; Kehrer-Sawatzki, H. Independent intrachromosomal recombination events underlie the pericentric inversions of chimpanzee and gorilla chromosomes homologous to human chromosome 16. Genome Res. 2005, 15, 1232–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, M.K.; Kim, C.-G.; Kitano, T.; Ferrell, R.E.; Kohara, Y.; Saitou, N. Nucleotide sequence comparison of a chromosome rearrangement on human chromosome 12 and the corresponding ape chromosomes. Cytogenet. Genome Res. 2005, 108, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Szamalek, J.M.; Goidts, V.; Searle, J.B.; Cooper, D.N.; Hameister, H.; Kehrer-Sawatzki, H. The chimpanzee-specific pericentric inversions that distinguish humans and chimpanzees have identical breakpoints in Pan troglodytes and Pan paniscus. Genomics 2006, 87, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrer-Sawatzki, H.; Szamalek, J.M.; Tänzer, S.; Platzer, M.; Hameister, H. Molecular characterization of the pericentric inversion of chimpanzee chromosome 11 homologous to human chromosome 9. Genomics 2005, 85, 542–550. [Google Scholar] [CrossRef]
- Dennis, M.Y.; Harshman, L.; Nelson, B.J.; Penn, O.; Cantsilieris, S.; Huddleston, J.; Antonacci, F.; Penewit, K.; Denman, L.; Raja, A.; et al. The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol. 2017, 1, 69. [Google Scholar] [CrossRef]
- Cheng, Z.; Ventura, M.; She, X.; Khaitovich, P.; Graves, T.; Osoegawa, K.; Church, D.; DeJong, P.; Wilson, R.K.; Paabo, S.; et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature 2005, 437, 88–93. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, M.; Kawahara, Y.; Nei, M. Genomic drift and copy number variation of sensory receptor genes in humans. Proc. Natl. Acad. Sci. USA 2007, 104, 20421–20426. [Google Scholar] [CrossRef] [Green Version]
- Fudenberg, G.; Pollard, K.S. Chromatin features constrain structural variation across evolutionary timescales. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, L.; Hormozdiari, F. TAD fusion score: Discovery and ranking the contribution of deletions to genome structure. Genome Biol. 2019, 20, 60. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.M.; de Manuel, M.; Marques-Bonet, T.; Castellano, S.; Andrés, A.M. The impact of genetic adaptation on chimpanzee subspecies differentiation. PLoS Genet. 2019, 15, e1008485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, R.R.; Kreitman, M.; Aguadé, M. A test of neutral molecular evolution based on nucleotide data. Genetics 1987, 116, 153–159. [Google Scholar]
- Yohn, C.T.; Jiang, Z.; Sean, D.; Karen, M.; Hayden, E.; Khaitovich, P.; Matthew, E.; Marla, J.; Eichler, Y. Lineage-Specific Expansions of Retroviral Insertions within the Genomes of African Great Apes but Not Humans and Orangutans. PLoS Biol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Chander, V.; Gibbs, R.A.; Sedlazeck, F.J. Evaluation of computational genotyping of structural variation for clinical diagnoses. Gigascience 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Lazar, N.H.; Nevonen, K.A.; O’Connell, B.; McCann, C.; O’Neill, R.J.; Green, R.E.; Meyer, T.J.; Okhovat, M.; Carbone, L. Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res. 2018, 28, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Ghavi-Helm, Y.; Jankowski, A.; Meiers, S.; Viales, R.R.; Korbel, J.O.; Furlong, E.E.M. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 2019, 51, 1272–1282. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huang, S.-C.; Glenn, S.; Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.-R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e24. [Google Scholar] [CrossRef] [Green Version]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; Consortium, G.; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monajemi, H.; Fontijn, R.D.; Pannekoek, H.; Horrevoets, A.J.G. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics 2002, 79, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Cui, Y.-H.; Han, Y.-H.; Fagerness, J.A.; Galloway, B.; Shen, Y.-C.; Kojima, T.; Uchiyama, M.; Faraone, S.V.; Tsuang, M.T. Association of SNPs and haplotypes in APOL1, 2 and 4 with schizophrenia. Schizophr. Res. 2008. [Google Scholar] [CrossRef] [Green Version]
- Balogh, A.; Toth, E.; Romero, R.; Parej, K.; Csala, D.; Szenasi, N.L.; Hajdu, I.; Juhasz, K.; Kovacs, A.F.; Meiri, H.; et al. Placental Galectins Are Key Players in Regulating the Maternal Adaptive Immune Response. Front. Immunol. 2019, 10, 1240. [Google Scholar] [CrossRef]
- Than, N.G.; Romero, R.; Goodman, M.; Weckle, A.; Xing, J.; Dong, Z.; Xu, Y.; Tarquini, F.; Szilagyi, A.; Gal, P.; et al. A primate subfamily of galectins expressed at the maternal-fetal interface that promote immune cell death. Proc. Natl. Acad. Sci. USA 2009, 106, 9731–9736. [Google Scholar] [CrossRef] [Green Version]
- Than, N.G.; Balogh, A.; Romero, R.; Kárpáti, E.; Erez, O.; Szilágyi, A.; Kovalszky, I.; Sammar, M.; Gizurarson, S.; Matkó, J.; et al. Placental Protein 13 (PP13)—A Placental Immunoregulatory Galectin Protecting Pregnancy. Front. Immunol. 2014, 5, 348. [Google Scholar] [CrossRef] [Green Version]
- Andres, A.M.; Dennis, M.Y.; Kretzschmar, W.W.; Cannons, J.L.; Lee-Lin, S.Q.; Hurle, B.; Comparative, N.; Program, S.; Schwartzberg, P.L.; Williamson, S.H.; et al. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet. 2010, 6, e1001157. [Google Scholar] [CrossRef] [Green Version]
- Wedenoja, S.; Yoshihara, M.; Teder, H.; Sariola, H.; Gissler, M.; Katayama, S.; Wedenoja, J.; Häkkinen, I.M.; Ezer, S.; Linder, N.; et al. Balancing Selection at HLA-G Modulates Fetal Survival, Preeclampsia and Human Birth Sex Ratio. BioRxiv 2019. [Google Scholar] [CrossRef]
- Tan, Z.; Shon, A.M.; Ober, C. Evidence of balancing selection at the HLA-G promoter region. Hum. Mol. Genet. 2005, 14, 3619–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | ENSEMBL ID | SV Type | Description |
|---|---|---|---|
| APOBR | ENSG00000184730 | deletion | Apolipoprotein B receptor |
| APOL1 | ENSG00000100342 | deletion | Apolipoprotein L1 |
| APOL4 * | ENSG00000100336 | deletion | Apolipoprotein L4 |
| ATP2A1 | ENSG00000196296 | deletion | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 |
| ATXN2L | ENSG00000168488 | deletion | Ataxin 2 like |
| CARD18 | ENSG00000255501 | deletion | Caspase recruitment domain family member 18 |
| CAST * | ENSG00000153113 | inversion | Calpastatin |
| CD19 | ENSG00000177455 | deletion | CD19 Molecule |
| CEACAM21 | ENSG00000007129 | deletion | CEA Cell Adhesion Molecule 21 |
| CFHR2 | ENSG00000080910 | deletion | Complement Factor H Related 2 |
| CFHR4 | ENSG00000134365 | deletion | Complement Factor H Related 4 |
| CLC | ENSG00000105205 | deletion | Charcot-Leyden crystal Galectin |
| CLN3 * | ENSG00000188603 | deletion | CLN3 Lysosomal/Endosomal Transmembrane Protein, Battenin |
| CMPK1 | ENSG00000162368 | deletion | Cytidine/Uridine Monophosphate Kinase 1 |
| CROCC | ENSG00000058453 | inversion | Ciliary Rootlet Coiled-Coil, Rootletin |
| CYP2C18 | ENSG00000108242 | deletion | Cytochrome P450 Family 2 Subfamily C Member 18 |
| DEFB128 | ENSG00000185982 | deletion | Defensin Beta 128 |
| EFCAB13 * | ENSG00000178852 | deletion | EF-Hand Calcium Binding Domain 13 |
| EIF3C * | ENSG00000184110 | deletion | Eukaryotic Translation Initiation Factor 3 Subunit C |
| IL18R1 * | ENSG00000115604 | inversion | Interleukin 18 Receptor 1 |
| IL1RL1 | ENSG00000115602 | inversion | Interleukin 1 Receptor Like 1 |
| IL27 | ENSG00000197272 | deletion | Interleukin 27 |
| IL36B | ENSG00000136696 | deletion | Interleukin 36B |
| IL37 | ENSG00000125571 | deletion | Interleukin 37 |
| KRTAP19-6 | ENSG00000186925 | deletion | Keratin Associated Protein 19-6 |
| KRTAP19-7 | ENSG00000244362 | deletion | Keratin Associated Protein 19-7 |
| LCN10 | ENSG00000187922 | deletion | Lipocalin 10 |
| LCN6 | ENSG00000267206 | deletion | Lipocalin 6 |
| LGALS14 | ENSG00000006659 | deletion | Galectin 14 |
| MERTK | ENSG00000153208 | deletion | MER Proto-Oncogene, Tyrosine Kinase |
| NPIPB8 * | ENSG00000255524 | deletion | Nuclear Pore Complex Interacting Protein Family Member B8 |
| NPIPB9 * | ENSG00000196993 | deletion | Nuclear Pore Complex Interacting Protein Family Member B9 |
| NUPR1 * | ENSG00000176046 | deletion | Nuclear Protein 1, Transcriptional Regulator |
| OBP2A | ENSG00000122136 | deletion | Odorant Binding Protein 2A |
| OR10H1 | ENSG00000186723 | deletion | Olfactory Receptor Family 10 Subfamily H Member 1 |
| OR10H5 | ENSG00000172519 | deletion | Olfactory Receptor Family 10 Subfamily H Member 5 |
| OR2T33 | ENSG00000177212 | deletion | Olfactory Receptor Family 2 Subfamily T Member 33 |
| OR6C2 | ENSG00000179695 | deletion | Olfactory Receptor Family 6 Subfamily C Member 2 |
| OR6C3 | ENSG00000205329 | deletion | Olfactory Receptor Family 6 Subfamily C Member 3 |
| OR6C65 | ENSG00000205328 | deletion | Olfactory Receptor Family 6 Subfamily C Member 65 |
| OR6C70 | ENSG00000184954 | deletion | Olfactory Receptor Family 6 Subfamily C Member 70 |
| OR6C75 | ENSG00000187857 | deletion | Olfactory Receptor Family 6 Subfamily C Member 75 |
| OR6C76 | ENSG00000185821 | deletion | Olfactory Receptor Family 6 Subfamily C Member 76 |
| POU6F2 | ENSG00000106536 | deletion | POU Class 6 Homeobox 2 |
| RABEP2 * | ENSG00000177548 | deletion | Rabaptin, RAB GTPase Binding Effector Protein 2 |
| RACK1 | ENSG00000204628 | inversion | Receptor For Activated C Kinase 1 |
| SGF29 * | ENSG00000176476 | deletion | SAGA Complex Associated Factor 29 |
| SH2B1 | ENSG00000178188 | deletion | SH2B Adaptor Protein 1 |
| SLC35G4 | ENSG00000236396 | deletion | Solute Carrier Family 35 Member G4 |
| SLCO1B3 * | ENSG00000111700 | inversion | Solute Carrier Organic Anion Transporter Family Member 1B3 |
| SULT1A1 * | ENSG00000196502 | deletion | Sulfotransferase Family 1A Member 1 |
| SULT1A2 | ENSG00000197165 | deletion | Sulfotransferase Family 1A Member 2 |
| TUFM | ENSG00000178952 | deletion | Tumor Protein P53 |
| YAE1D1 | ENSG00000241127 | deletion | YAE1 Maturation Factor Of ABCE1 |
| AC011604.2 | ENSG00000257046 | inversion | Uncharacterized |
| AL355987.1 | ENSG00000204003 | deletion | Uncharacterized |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto, D.C.; Shew, C.; Mastoras, M.; Schmidt, J.M.; Sahasrabudhe, R.; Kaya, G.; Andrés, A.M.; Dennis, M.Y. Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing. Genes 2020, 11, 276. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030276
Soto DC, Shew C, Mastoras M, Schmidt JM, Sahasrabudhe R, Kaya G, Andrés AM, Dennis MY. Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing. Genes. 2020; 11(3):276. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030276
Chicago/Turabian StyleSoto, Daniela C., Colin Shew, Mira Mastoras, Joshua M. Schmidt, Ruta Sahasrabudhe, Gulhan Kaya, Aida M. Andrés, and Megan Y. Dennis. 2020. "Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing" Genes 11, no. 3: 276. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030276