Transcriptomic Profiling of DAF-7/TGFβ Pathway Mutants in C. elegans


1
Department of Cell Development and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA
2
Department of Genetics, University of Alabama at Birmingham, Birmingham, AL 35294, USA
3
Department of Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL 35294, USA
*
Author to whom correspondence should be addressed.
Genes 2020, 11(3), 288; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030288
Submission received: 16 January 2020
/
Revised: 26 February 2020
/
Accepted: 3 March 2020
/
Published: 9 March 2020
(This article belongs to the Special Issue C. elegans - A tribute to the legendary worm)
Abstract
:The transforming growth factor beta superfamily encompasses a large family of ligands that are well conserved across many organisms. They are regulators of a number of physiological and pathological processes. The model nematode Caenorhabditis elegans has been instrumental in identifying key components of the transforming growth factor beta (TGFβ) pathway. In C. elegans, the TGFβ homolog DAF-7 signals through the DAF-1 Type I and DAF-4 Type II receptors to phosphorylate downstream R-SMADs DAF-8 and DAF-14. These R-SMADs translocate into the nucleus to inhibit Co-SMAD DAF-3. Many of the roles of the canonical DAF-7 pathway, involving both DAF-1 and DAF-3, have been identified using targeted genetic studies. Few have assessed the global transcriptomic changes in response to these genes, especially in adult animals. In this study, we performed RNA sequencing on wild type, daf-1, and daf-1; daf-3 adult hermaphrodites. To assess the overall trends of the data, we identified differentially expressed genes (DEGs) and performed gene ontology analysis to identify the types of downstream genes that are differentially expressed. Hierarchical clustering showed that the daf-1; daf-3 double mutants are transcriptionally more similar to wild type than daf-1 mutants. Analysis of the DEGs showed a disproportionally high number of genes whose expression is increased in daf-1 mutants, suggesting that DAF-1 acts as a general repressor of gene expression in wild type animals. Gene ontology analysis of the DEGs produced many significantly enriched terms, including Molting Cycle, Response to Topologically Incorrect Protein, and Response to Biotic Stimulus. Understanding the direct and indirect targets of the DAF-7 TGFβ pathway through this RNA-seq dataset can provide insight into novel roles of the multifunctional signaling pathway, as well as identify novel genes that may participate in previously reported functions of TGFβ signaling.
1. Introduction
Transforming growth factor beta (TGFβ) is the prototype of the large family of secreted ligands that act through downstream receptors to modulate diverse biological functions, such as embryological development, adult stem cell differentiation, immune regulation, wound healing, inflammation, and cancer [1,2,3,4,5,6]. The functionality of this family of proteins is highly conserved among many organisms, including human, mice, nematodes, and flies [7,8,9]. Canonically, TGFβ signals as homo- and heterodimers to activate serine/threonine kinase receptors that act as heterotetramers to phosphorylate downstream receptor-regulated SMADs (R-SMADs). The activated R-SMADs then translocate into the nucleus to regulate the transcription of a wide array of downstream targets [10]. Studies in the past three decades have provided great insight into the multifunctionality of the TGFβ pathway in affecting developmental and pathologic mechanisms, revealing different, and sometimes opposite, responses to TGFβ. For example, while TGFβ has been shown to inhibit the growth of many cell types, such as epithelial [11,12], hematopoietic [13,14], and glial cells [15,16], it can also stimulate the growth of other cell types, such as chondrocytes and osteoblasts [17,18]. These studies unveil the complexities of this pathway and suggest that identification of key effectors of the pathway is crucial to teasing out the mechanisms underlying the differential effects of TGFβ.
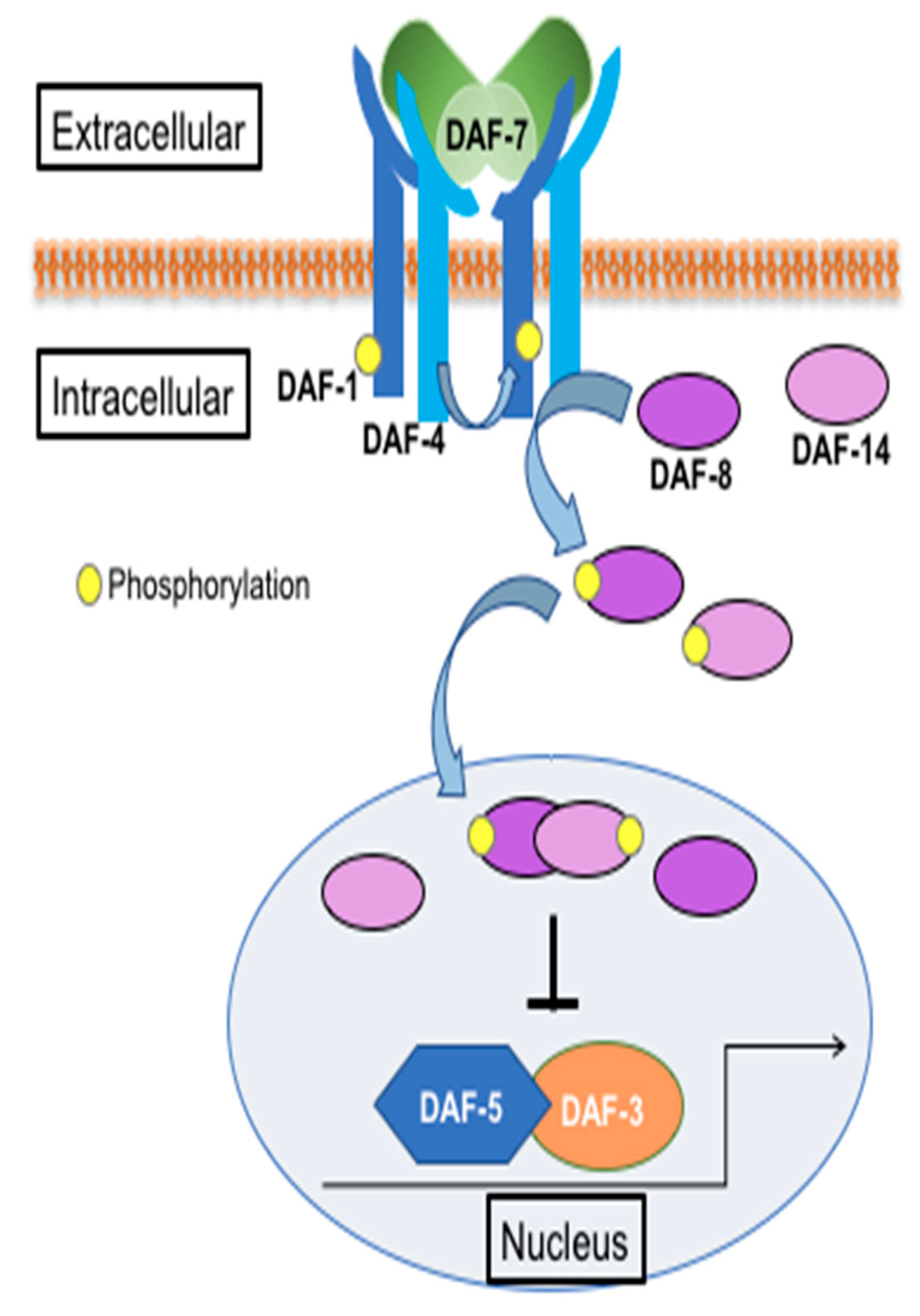
Genetic studies in Caenorhabditis elegans have been instrumental in the identification of key TGFβ pathway components. In C. elegans, DAF-7 encodes a TGFβ homolog and is known primarily for its role in regulating dauer formation [19]. DAF-7 is secreted from the ASI sensory neurons in response to environmental cues, such as food, pheromones, and pathogens [20,21]. It acts as a homodimer to bind the DAF-1 Type I and DAF-4 Type II ser/thr kinase receptors. These receptors function as heterotetramers to phosphorylate downstream R-SMADs DAF-8 and DAF-14. The activated SMADs translocate to the nucleus to suppress the Co-SMAD DAF-3 (Figure 1) [8]. The daf genes were initially identified for their roles in dauer formation. They are divided into two categories: dauer constitutive (e.g., daf-7, daf-1, daf-4, daf-8, and daf-14) and dauer defective (daf-3). Genetic crosses of dauer constitutive and dauer defective mutants result in double mutants, such as daf-1; daf-3, that are dauer defective, suggesting that daf-3 is epistatic to daf-1. This relationship has been observed in many other functions of the pathway, some of which may be conserved in higher animals, including humans.
In addition to dauer formation, DAF-7 in C. elegans has been shown to play a role in regulating the expression of AMPA-type glutamate receptor GLR-1 [22], the expression of chemoreceptors in amphid neurons [23], the expression of genes in the distal tip cell to modulate the germline stem cell niche [24,25], and the production of prostaglandins that are important for sperm guidance [26]. These studies illustrate that, similar to mammalian systems, C. elegans TGFβ regulates a wide array of biological functions. Understanding the direct and indirect targets of this pathway may uncover novel functions and regulatory mechanisms that are translatable to mammalian systems.
Many of the known targets downstream of DAF-1 and DAF-3 have been identified via direct genetic studies or forward genetic screens. Few studies have characterized the transcriptional profiles of adult TGFβ mutants [27]. In this study, we performed RNA sequencing on wild-type, daf-1, and daf-1; daf-3 mutants. We looked at the general trends of the RNA sequencing data, focusing on the genes that are differentially altered by daf-1 and daf-3. The datasets provided in this study are some of the first to be generated by RNA sequencing of the DAF-7 TGFβ pathway mutants. They provide a tool that can facilitate future work in identifying DAF-3 dependent and independent functions and downstream factors of the pathway in C. elegans.
2. Materials and Methods
2.1. C. elegans Strains and Maintenance
C. elegans strains Bristol N2 (wild type), daf-1(m40), and daf-3(mgDf90) were ordered from the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs. The daf-1(m40); daf-3(mgDf90) double mutant strain was generated through genetic crosses. C. elegans strains were maintained in NA22 Escherichia coli bacteria on nematode growth media (NGM) plates at 16 °C or 25 °C.
To grow worms for RNA isolation, worms were first synchronized by the bleaching technique [28]. Briefly, gravid worms were recovered from ten 10 cm plates by washing with M9 buffer. Worm pellets were washed with M9 buffer until the buffer was clear of bacteria. The resulting worm pellet was exposed to an alkaline hypochlorite solution (1 mg NaOH, 25 mL sodium hypochlorite, and 74 mL H2O) and vortexed for 2 min. The mixture was centrifuged up to 1000× g for 5 s. The resulting pellet contained a white layer (eggs) and a yellow layer (intact gravid worms). The supernatant was discarded, and the process was repeated with fresh alkaline hypochlorite solution until the pellet was mostly white. The reaction was terminated by washing 5–6 times with M9 buffer. The resulting white pellet was resuspended in 6 mL of S. media and agitated overnight at 16 °C on a nutator to allow the eggs to hatch into L1 larva. The synchronized L1 larva were plated on 15 cm NGM plates and supplemented with thick NA22 E. coli. The synchronized worms were grown at 16 °C until the L4 stage and shifted to 25 °C for 24 h before harvesting with M9 buffer. The worms were washed with M9 buffer until the supernatant was clear of bacteria, and the worm pellets were stored in 250 mg aliquots at −80 °C.
2.2. RNA Isolation
250 mg of frozen, synchronized worms were homogenized with a Bullet Blender 5 (Next Advance, Troy, NY, USA) and 0.5 mm zirconium beads. Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) following the kit’s protocol.
2.3. Next-Generation Sequencing on Illumina Platforms
Sequencing of the total RNA was performed at the University of Alabama at Birmingham (UAB) Heflin Center for Genomic Science Core Laboratories. mRNA-sequencing was performed on the Illumina HiSeq2500 using the latest versions of the sequencing reagents and flow cells providing up to 300 Gb of sequence information per flow cell. Briefly, the quality of the total RNA was assessed using the Agilent 2100 Bioanalyzer followed by 2 rounds of poly A+ selection and conversion to cDNA. We used the TruSeq library generation kits as per the manufacturer’s instructions (Illumina, San Diego, CA, USA). Library construction consists of random fragmentation of the polyA mRNA, followed by cDNA production using random primers. The ends of the cDNA were repaired and A-tailed, and adaptors ligated for indexing (up to 12 different barcodes per lane) during the sequencing runs. The cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for library quantitation (Kapa Biosystems, Woburn, MA) prior to cluster generation. Clusters were generated to yield approximately 725–825 K clusters/mm2. Cluster density and quality were determined during the run after the first base addition parameters were assessed. We ran paired end 2 × 50 bp sequencing runs to align the cDNA sequences to the reference genome (Table S1).
2.4. Data Assessment
The raw FASTQ reads were first trimmed using TrimGalore! Version 0.4.0 to remove any primer adapter contamination. TopHat version 2.0.12 was then used to align the trimmed FASTQ reads to the Ensembl reference genome WBcel235.ceWS240 Release 76 (www.ensembl.org) using the short read aligner Bowtie version 2.2.3 [29,30,31]. Cufflinks version 2.2.1 with default parameters used the aligned reads from TopHat to assemble transcripts, estimate their abundances, and test for differential expression and regulation [31,32]. Cuffmerge, which is part of Cufflinks that merged the assembled transcripts to a reference annotation. Finally, Cuffdiff found significant changes in transcript expression. When looking for genes with statistically significant changes in absolute expression, Cuffdiff uses a t-test to calculate the p values of the observed changes and Benjamini-Hochberg multiple testing correction to calculate the q values.
2.5. Data Visualization and Gene Ontology
Principle component analysis (PCA) analysis and heatmapping of the data were performed using the fragments per kilobase of exon model per million reads mapped (FPKM) values of the individual samples in Rstudio (Version 1.1.456) [33]. PCA was calculated by inputting the normalized FPKM values into the prcompt command with Scale = TRUE and plotted using ggplot2 [34]. The heatmap was generated using heatmap.2 of z transformed FPKM values [35]. For gene ontology, differentially expressed genes (DEGs) in the daf-1 vs. N2 dataset with ≥2 fold difference, q < 0.05 were first identified. These genes were then compared to the DEGs in the daf-1 vs. daf-1; daf-3 dataset with ≥2 fold difference, p < 0.05, q < 0.05. Only genes that were increased or decreased in daf-1 when compared to both N2 and daf-1; daf-3 were used for gene ontology analysis. The list of identified genes was entered into the enrichment analysis tool on wormbase.org [36]. The gene ontology terms were recorded, and parent child terms were manually identified and noted in the data tables.
2.6. Data Validation by qRT-PCR
The same RNA used for RNA-seq was used for quantitative real-time polymerase chain reaction (qRT-PCR) analysis. cDNAs were synthesized from 1µg of RNA using the SuperScript IV Reverse Transcriptase (Invitrogen) kit and random hexamers following the provided protocol. Real-time PCR was performed in the Bio-Rad CFX Connect Real-Time System with Syber Green master mix (Bio-Rad, Hercules, CA, USA) following the provided protocol. Each sample was run in triplicate. Three biological samples were run for N2 (wild-type), daf-1(m40), and daf-1(m40); daf-3(mgDf90) double mutants. The PCR protocol used was: 95 °C for 2 min; 40 cycles of 95 °C for 5 s and 60 °C for 30 s. The primers used are listed in Table S2. The relative expression fold change was calculated in the CFX manager software system (Bio-Rad) using the 2^−ddCT method. cdc-42 was used as a control.
3. Results
3.1. Overall Trends of the Wild-Type and TGFβ Mutant Transcriptomes
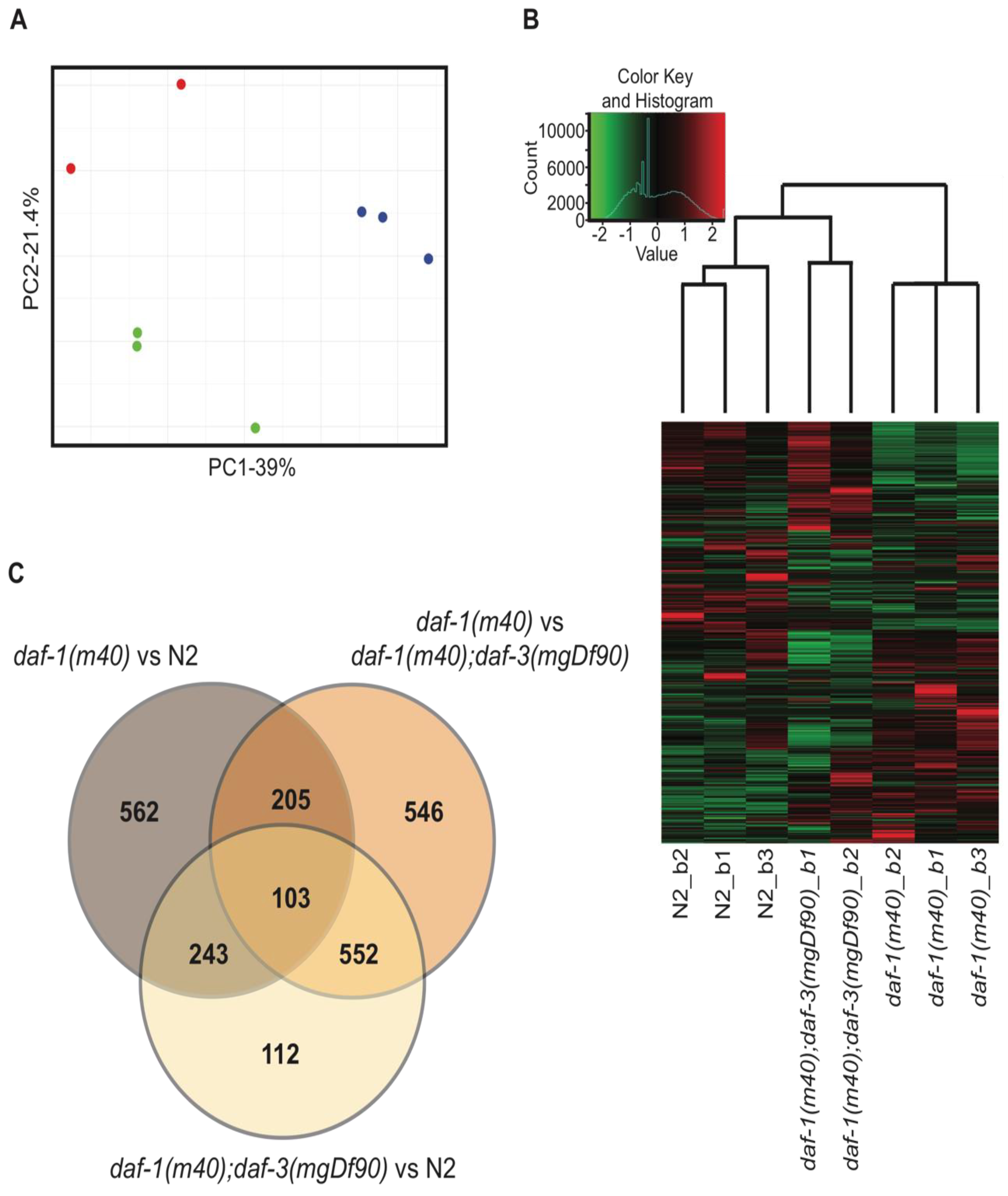
We extracted total RNA from N2, daf-1, and daf-1; daf-3 adult hermaphrodites. Three biological replicates (i.e., batches, denoted as b1-b3) per genotype were processed for RNA sequencing. PCA using the FPKM values of all 9 samples showed that daf-1; daf-3 batch 3 was an extreme outlier (Figure S1). This sample was therefore removed from further analyses. PCA of the remaining 8 samples showed close clustering within replicates of the same genotype and a high degree of variance between genotypes (Figure 2A). Hierarchical clustering in the heatmap further confirmed that replicates of the same genotype were more similar to each other than to samples of other genotypes (Figure 2B). It was of interest that daf-1; daf-3 double mutants clustered more closely with wild-type than daf-1 mutants. This finding coincided with many functional studies showing that daf-3 suppresses the phenotypes of daf-1 single mutants, suggesting that that daf-1; daf-3 double mutants are also functionally more similar to wild-type than daf-1 animals.
We used Cufflinks to identify DEGs between the sequenced genotypes. Only genes that showed a ≥ 2-fold difference at q < 0.05 were identified for downstream analysis. 1113 DEGs passed these criteria in the daf-1 vs. N2 dataset and 1406 DEGs were identified in the daf-1 vs. daf-1; daf-3 dataset (Figure 2C, Tables S3 and S4). Of these DEGs, only 308 genes overlapped between the two datasets. This suggested that while daf-1 and daf-3 each contributed to wide spread changes in the transcriptome of the C. elegans, only a small fraction of downstream targets are regulated by both daf-1 and daf-3.
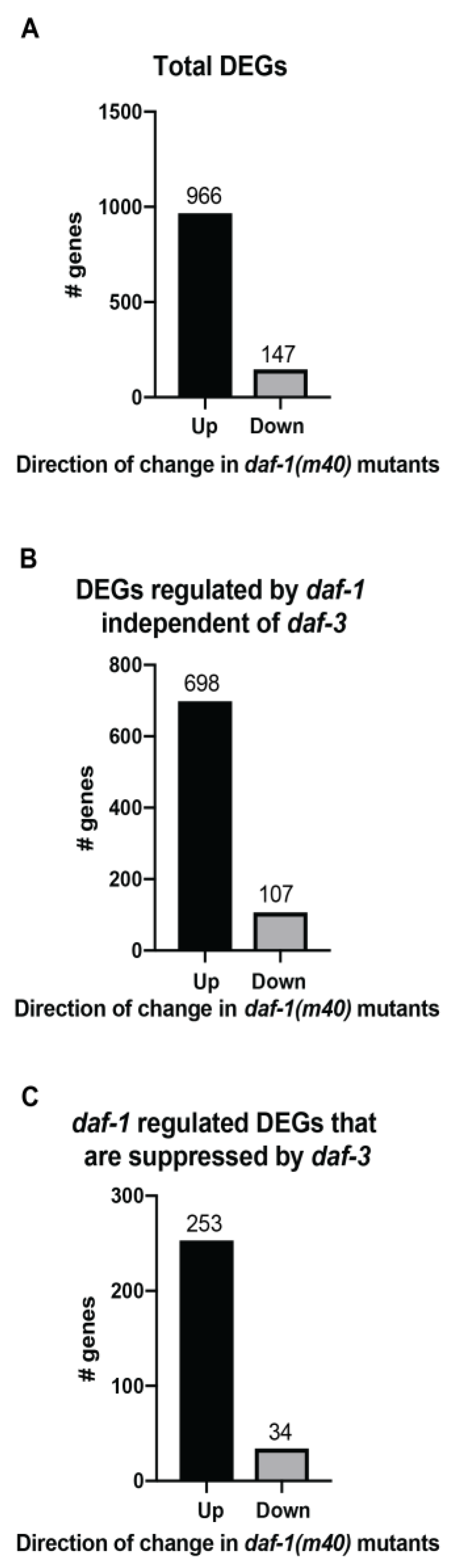
To understand how daf-1 and daf-3 regulated their downstream targets, we assessed the number of up and down regulated genes in each dataset. Of the 1113 DEGs in the daf-1 vs. N2 dataset, 966 were increased in daf-1 mutants (Figure 3A), suggesting that daf-1 acts as a general repressor of gene expression in wild-type animals. We dissected the data further into genes that were regulated by daf-1 independent of daf-3 (i.e., genes whose expression is altered in the daf-1 vs. N2 dataset, but not in the daf-1 vs. daf-1; daf-3 dataset, 805 genes total) and genes that were differentially regulated by daf-1 and daf-3 (i.e., genes that were found to be increased in daf-1 mutants when compared to wild-type and decreased in daf-1; daf-3 double mutants when compared to daf-1, 287 genes total).We found that in both groups, the majority of genes were upregulated in daf-1 mutants (Figure 3B,C). These results suggested that in the context of the daf-1 and daf-3 signaling pathway, daf-3 was responsible for driving the transcription of the bulk of their downstream targets, while daf-1 worked to decrease their expression. Functional studies of this pathway have emphasized the role of daf-1 and daf-3 working in sequence to regulate the bulk of the reported phenotypes. However, the transcriptomics studies presented here highlight that daf-1 and daf-3 share only a small percentage of downstream targets.
3.2. Functional Annotation of the Transcriptomic Data
Our goal in this study was to understand the function of genes downstream of the canonical DAF-7 TGFβ pathway, which involves both daf-1 and daf-3. We performed GO enrichment analysis using the 287 genes that are differentially regulated by daf-1 and daf-3 to probe for the possible functions of these downstream genes. Table 1 contains the list of GO terms that were significantly enriched (q < 0.05). Analysis of the 34 genes that were downregulated in daf-1 mutants showed an enrichment of the biological process Response to Biotic Stimulus. The 253 upregulated genes mapped to many more GO terms in all three major GO categories. Significantly enriched terms in the biological processes included ‘Response to Topologically Incorrect Protein Folding’, ‘Response to Biotic Stimulus’, and ‘Molting Process’. Significantly enriched parent terms under cellular components included Collagen Trimer, Extracellular Region, and Membrane. Significantly enriched terms for molecular function included Structural Constituent of Cuticle, Passive Transmembrane Transporter Activity, and Peptidase Activity. Collagen Trimer and Structural Constituent of Cuticle were two of the most highly enriched terms, at 26- and 28-fold enrichment respectively. This reflected the known role of the DAf-7 TGFβ pathway in modulating the progression of larval development, which also involves the regulation of cuticle components.
We next focused on biological process terms to tease out the type of genes annotated to each GO term, as proof of principle (Table 2). For all of the genes listed in Table 2, their functions or predicted functions were identified using WormBase. For the genes in the Molting Process, dpy-13, rol-6, sqt-1, and sqt-2 are all reportedly associated with components of the cuticle and collagen. nas-30 is a metalloproteinase and is predicted to play a role in the hatching process. The term response to Biotic Stimulus were associated with genes both up- and down- regulated in daf-1 mutants. Of the up-regulated genes, sodh-1, clec-61, and math-38 are associated with defense to gram positive bacterium. irg-1 and F20G2.5 participate in defense to gram negative bacterium and the innate response. fir-7 is a fungus induced protein, and nlp-30 is expressed in the hypodermis and responds to both bacterial and fungal infections. Of the down-regulated genes, ilys-5 is predicted to have lysozyme activity, clec-218 has predicted carbohydrate binding activity and responds to gram-positive bacteria. lys-4 has predicted lysozyme activity as well as defense to gram positive bacteria. All of the genes annotated to the response to Topologically Incorrect Protein Folding either participated or were predicted to participate in the endoplasmic reticulum unfolded protein response. abu-1, abu-8, abu-10, and abu-11 are members of a family of genes called Activated in Blocked Unfolded Protein Response. col-109 is a component of the cuticle, pqn-74 has predicted chitin binding activity, and clec-67 has predicted carbohydrate binding activity. Fipr-27 is a member of the fungus-induced protein family.
3.3. Validation of Select RNA-Sequencing Genes by Quantitative Real-Time PCR
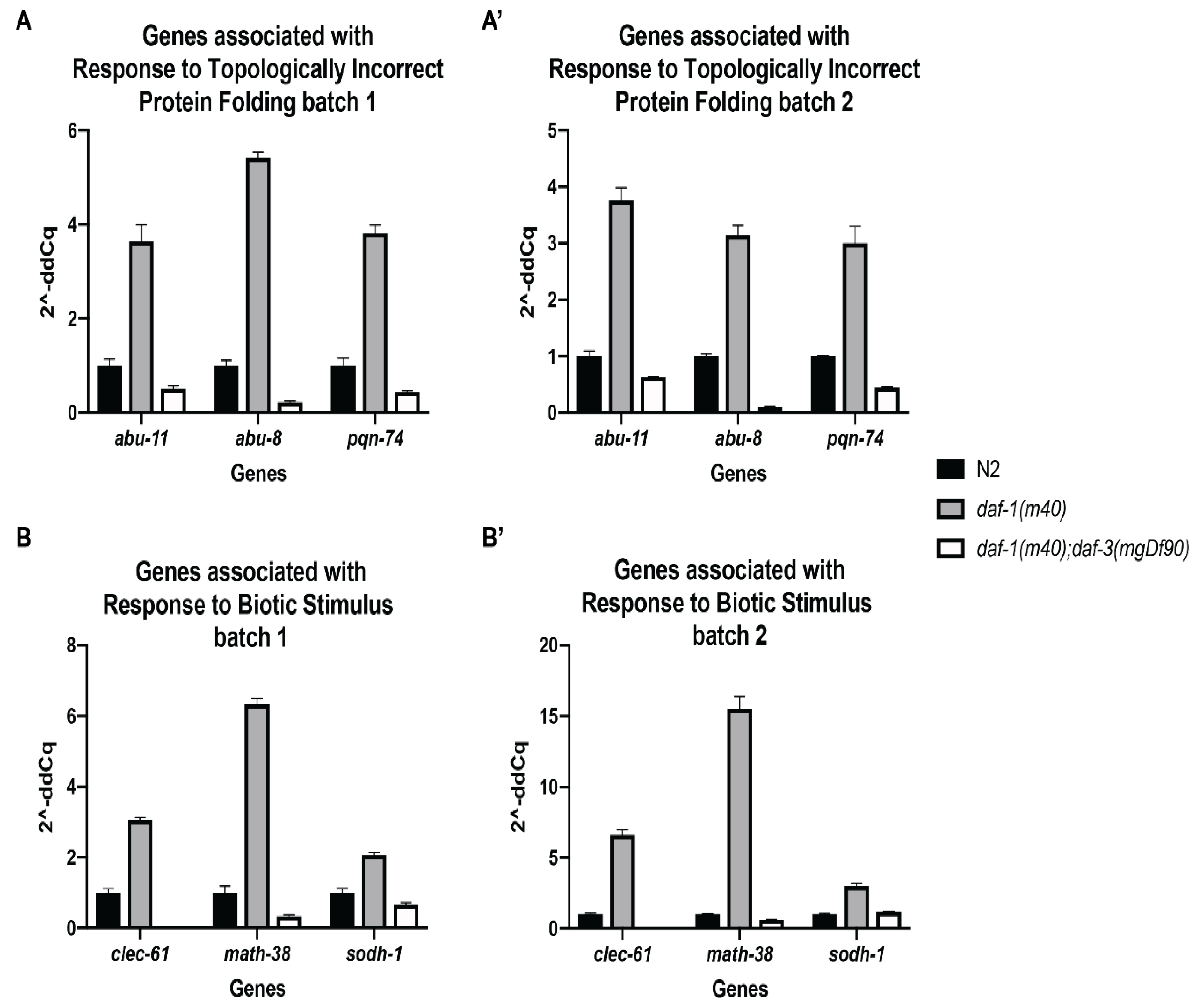
To validate the gene expression changes observed in the RNA-seq data, we performed quantitative real-time PCR on three genes from both Response to Topologically Incorrect Protein Folding and Response to Biotic Stimulus. We used the same RNA that was submitted for RNA sequencing for the qPCR analysis. RNA from three biological replicates (i.e., batches) of N2 wild-type, daf-1 single mutant, and daf-1; daf-3 double mutant worms were assessed. cdc-42 was selected as the only reference gene used for the validation study. We chose this gene because cdc-42 has been a reliable reference gene used in many genetic backgrounds, and its FPKM value was approximately equal across all of our samples. Figure 4 depicts the expression fold change seen in samples from batch 1 and batch 2, corresponding to the batch numbers from the RNA-seq samples. The genes abu-11, abu-8, and pqn-74 were selected from the Response to Topologically Incorrect Protein Folding GO term. The expression of these genes was increased in daf-1 mutants compared to wild type, and this increase was suppressed in the daf-1; daf-3 double mutants (Figure 4A,A’). A similar trend is seen with the Response to Biotic Stimulus genes clec-61, math-38, and sodh-1 (Figure 4B,B’). qPCR using RNA from batch 3 of N2, daf-1, and daf-1; daf-3 showed an increase in expression of all six genes in daf-1. However, a corresponding suppression of expression in the daf-1; daf-3 sample was not observed for abu-8, pqn-74, and sodh-1 [37], underscoring the possibility that this sample is a true outlier.
4. Discussion
TGFβ signaling is ubiquitously found to regulate many physiological and pathological processes and is highly conserved among many organisms [7,8,9]. In C. elegans, DAF-7 is a TGFβ homolog that is secreted by ASI sensory neurons in response to environmental cues, such as pheromones and food. The canonical DAF-7 TGFβ pathway signals through the DAF-1 Type I and DAF-4 Type II receptors to downstream DAF-8 and DAF-14. These R-SMADs translocate into the nucleus to inhibit the Co-SMAD DAF-3 [8,20]. This pathway has been heavily studied for its role in dauer formation and has also been implicated in other processes, such as germline development, neuronal gene expression, and fat metabolism [19,23,24,38]. While these studies have provided insight into downstream genes that are regulated by this canonical signaling mechanism, few studies have investigated the global transcriptomic profile changes that are mediated by TGFβ signaling, especially in the adult worm [27]. In this study, we performed RNA sequencing on N2, daf-1, and daf-1; daf-3 adult hermaphrodites. We reported the general trends found in the sequencing data and focused on characterizing the transcriptomic profile of genes that are differentially regulated by both daf-1 and daf-3. These genes reflect pathways in which increases or decreases in gene expression in response to daf-1 are suppressed by daf-3.
Many studies on the DAF-7 TGFβ pathway reinforce the currently accepted model between daf-3 and its upstream regulators, in which a phenotypic or genotypic change observed in daf-1 mutants is suppressed in daf-1; daf-3 double mutants, suggesting that daf-3 is epistatic to daf-1 [24,25,26,38]. These findings magnify the canonical DAF-7 signaling pathway and suggest that daf-1; daf-3 double mutant animals are more similar to wild type animals than to daf-1 mutants. PCA and hierarchical clustering of our RNA-seq data show that daf-1; daf-3 clusters closer with N2 than daf-1, supporting the findings in the functional studies. However, a comparison of the DEGs between daf-1 versus wild type and daf-1 versus daf-1; daf-3 identified only a small fraction of overlapping genes, suggesting that of the downstream genes affected by daf-1 and daf-3 individually, only a small fraction are co-regulated by both. This also highlights the broader functions of daf-1 and daf-3 outside of this canonical TGFβ signaling pathway, though we focused on co-regulated genes that are differentially regulated by daf-1 and daf-3 and not those regulated by daf-1 and daf-3 independently.
The aim of this study was to assess the profile of genes downstream of the canonical DAF-7 TGFβ pathway. Therefore, we focused the downstream GO analysis on the genes that are differentially regulated by daf-1 and daf-3. The enriched GO terms reflect functions of the canonical pathway that have been shown extensively through other genetic and functional studies, as well as novel functions of the pathway that have not been well studied in C. elegans. For example, the canonical DAF-7 TGFβ pathway is known for its role in dauer formation. Dauer arrest is a decision made during the L1-L2 larval stages, where an animal changes its metabolism and morphology to survive harsh environmental conditions. daf-7, daf-1, daf-14, and daf-8 mutants are all prone to dauer formation, a phenotype that is suppressed by daf-3. Most mutant alleles are temperature sensitive and must be maintained at a lower permissive temperature until the L4 larval stage to prevent dauer formation [39,40]. Dauering is a process that extensively changes the development and morphology of the animal. Correspondingly, our data showed enrichment of genes involved in the Molting Cycle, as well as the Cuticle and Collagen Components.
The canonical DAF-7 pathway has also been shown to modulate gene expression and worm behavior in response to environmental stimuli. However, few studies have looked at the mechanisms by which the canonical pathway achieves these functions. DAF-7 has been demonstrated to affect the expression of olfactory chemoreceptors on chemosensory neurons, such as the ASI, and glutamate receptor GLR-1 in the ventral nerve cord [22,23]. It has also been shown to respond to pathogenic bacteria, Pseudomonas aeruginosa, in addition to other cues such as food, temperature, and pheromones [19,21]. A closer inspection of the genes involved in Passive Transmembrane Transporter activity showed many predicted ion channels that are expressed in neurons and neurotransmitter transporters, including dat-1, unc-8, unc-38, and twk-40. Moreover, the GO term Response to Biotic Stimulus contained mainly genes that help the organism respond to both gram-negative and gram-positive bacteria, as well as fungi (see Table 2). These GO terms, along with their associated genes, suggest that the canonical DAF-7 TGFβ pathway may play a significant regulatory role in affecting C. elegans chemosensation as well as response to pathogenic versus beneficial organisms in their surroundings.
Some of the GO terms that were identified in this study are not associated with any reported roles of the DAF-7 pathway, such as Response to Topologically Incorrect Protein. However, TGFβ has been reported to interact with the unfolded protein response in other model systems, such as hepatic stellate cells and corneal endothelial cells, suggesting this function is evolutionarily conserved [41,42]. Regardless of the novelty of the identified GO terms, the DEGs generated from these datasets provide specific gene targets that can be further studied to provide insight into the multifunctional TGFβ pathway.
A limitation of transcriptome studies and the ensuing gene ontology analysis hinges on the robustness of the annotations of the transcriptome database. It is important to keep in mind that as targeted genetic studies elucidate the function of specific genes, the gene ontology analysis will also evolve. In the current study, the presence of expected GO terms, in addition to novel GO categories, suggests that the ontologies are largely on track. However, certain genes, such as the abu/pqn genes, while annotated as Response to Incorrect Protein folding, have been reported to function as pharyngeal cuticle proteins [43]. Furthermore, while precautions have been taken to synchronize the samples to minimize the effect of the developmental stage on differential gene expression, it should still be noted that, although the worms were developmentally synchronized, development remains a potential confounding factor in transcriptomic studies. However, qPCR assessment of the selected genes provides validation for the trends seen in our RNA-seq data. Furthermore, we want to emphasize that this study provides a bird’s eye view of the transcriptome. The DEGs identified in this study include those with low FPKM values, though their differential expressions were greater than 2 fold. We retained this subset of genes in order to decrease the likelihood of excluding potentially significant genes, though small subsets of false positive results may also be included. This study focused mainly on the functions of genes that act downstream of both daf-1 and daf-3. The datasets generated in this study will facilitate future investigations into the roles of both the canonical DAF-7 TGFβ pathway as well as genes that are regulated by daf-1 independent of daf-3. While RNA-seq of the daf-3 single mutants was not included in this study, the differential gene expression provided in the daf-1 vs. daf-1; daf-3 and N2 vs. daf-1;daf-3 (Table S5) tables offers a glimpse of the genes that may be regulated by daf-3 independent of daf-1. Understanding the genes that are downstream of these TGFβ pathway components provides insight into the complexity of this broadly relevant signaling pathway and stimulates the development of new hypotheses regarding its roles in physiologic and pathologic processes.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4425/11/3/288/s1. Figure S1: PCA plot of all 9 samples. Table S1: Summary of RNA sequencing analysis. Table S2: List of primers used in qPCR validation. Table S3: List of differentially expressed genes between N2 and daf-1(m40). Table S4: List of differentially expressed genes between daf-1(m40) and daf-1(m40); daf-3(mgDf90). Table S5: List of differentially expressed genes between N2 and daf-1(m40); daf-3(mgDf90).
Author Contributions
M.H. and R.A.S. contributed to the conceptualization of the manuscript. M.H., J.K.P., D.C., M.A.M. and R.A.S. all participated in data analysis, including data acquisition, visualization, and validation. The original draft was prepared by M.H., with extensive review and editing by M.H. and R.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the NIH (R01 GM118361 to M.A.M. and J.K.P., and F30HD094446 to M.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
We thank the NIH supported Caenorhabditis Genetics Center for the worm strains (P40 OD010440). We thank Michael Crowley, David Crossman, and the UAB Heflin Center for Genomic Science Core Laboratories for performing RNA-seq and bioinformatics analyses (CA013148 Comprehensive Cancer Center Support Grant and AI027767 Center for AIDS Research). We thank Sarah Peters for providing help with real-time PCR. We thank Anita Hjelmeland’s lab, especially Catherine Libby, for lending their real-time PCR machine and reagents.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Colak, S.; Ten Dijke, P. Targeting TGF-beta signaling in cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-beta family in wound healing, burns and scarring: A review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the immune response by TGF-beta: From conception to autoimmunity and infection. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watabe, T.; Miyazono, K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009, 19, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. TGF-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Bottinger, E.P.; Letterio, J.J.; Roberts, A.B. Biology of TGF-beta in knockout and transgenic mouse models. Kidney Int. 1997, 51, 1355–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumienny, T.L.; Savage-Dunn, C. TGF-beta signaling in C. elegans. WormBook 2013, 1–34. [Google Scholar] [CrossRef]
- Upadhyay, A.; Moss-Taylor, L.; Kim, M.J.; Ghosh, A.C.; O’Connor, M.B. TGF-beta family signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, A.; Howe, P.H. The tale of transforming growth factor-beta (TGFbeta) signaling: A soigne enigma. IUBMB Life 2009, 61, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Mulder, K.M. Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Rev. 2000, 11, 23–35. [Google Scholar] [CrossRef]
- Petritsch, C.; Beug, H.; Balmain, A.; Oft, M. TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 2000, 14, 3093–3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderberg, S.S.; Karlsson, G.; Karlsson, S. Complex and context dependent regulation of hematopoiesis by TGF-beta superfamily signaling. Ann. NY Acad. Sci. 2009, 1176, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Scandura, J.M.; Boccuni, P.; Massague, J.; Nimer, S.D. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 15231–15236. [Google Scholar] [CrossRef] [Green Version]
- Vergeli, M.; mazzanti, B.; Ballerini, C.; Gran, B.; Amaducci, L.; Massacesi, L. Transforming growth factor-beta 1 inhibits the proliferation of rat astrocytes induced by serum and growth factors. J. Neurosci. Res. 1995, 40, 127–133. [Google Scholar] [CrossRef]
- Lindholm, D.; Castren, E.; Kiefer, R.; Zafra, F.; Thoenen, H. Transforming growth factor-beta 1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. J. Cell Biol. 1992, 117, 395–400. [Google Scholar] [CrossRef]
- Seo, H.S.; Serra, R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 2007, 310, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.; Lim, C.S.; Johnsen, R.; Albert, P.S.; Pilgrim, D.; Riddle, D.L. Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science 1996, 274, 1389–1391. [Google Scholar] [CrossRef]
- Schackwitz, W.S.; Inoue, T.; Thomas, J.H. Chemosensory neurons function in parallel to mediate a pheromone response in C. elegans. Neuron 1996, 17, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Meisel, J.D.; Panda, O.; Mahanti, P.; Schroeder, F.C.; Kim, D.H. Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans. Cell 2014, 159, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGehee, A.M.; Moss, B.J.; Juo, P. The DAF-7/TGF-beta signaling pathway regulates abundance of the Caenorhabditis elegans glutamate receptor GLR-1. Mol. Cell Neurosci. 2015, 67, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, K.M.; Sarafi-Reinach, T.R.; Horne, J.G.; Saffer, A.M.; Sengupta, P. The DAF-7 TGF-beta signaling pathway regulates chemosensory receptor gene expression in C. elegans. Genes Dev. 2002, 16, 3061–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalfo, D.; Michaelson, D.; Hubbard, E.J. Sensory regulation of the C. elegans germline through TGF-beta-dependent signaling in the niche. Curr. Biol. 2012, 22, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Pekar, O.; Ow, M.C.; Hui, K.Y.; Noyes, M.B.; Hall, S.E.; Hubbard, E.J.A. Linking the environment, DAF-7/TGFbeta signaling and LAG-2/DSL ligand expression in the germline stem cell niche. Development 2017, 144, 2896–2906. [Google Scholar] [CrossRef] [Green Version]
- McKnight, K.; Hoang, H.D.; Prasain, J.K.; Brown, N.; Vibbert, J.; Hollister, K.A.; Moore, R.; Ragains, J.R.; Reese, J.; Miller, M.A. Neurosensory perception of environmental cues modulates sperm motility critical for fertilization. Science 2014, 344, 754–757. [Google Scholar] [CrossRef] [Green Version]
- Shaw, W.M.; Luo, S.; Landis, J.; Ashraf, J.; Murphy, C.T. The C. elegans TGF-beta Dauer pathway regulates longevity via insulin signaling. Curr. Biol. 2007, 17, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Porta-de-la-Riva, M.; Fontrodona, L.; Villanueva, A.; Ceron, J. Basic Caenorhabditis elegans methods: Synchronization and observation. J. Vis. Exp. 2012, e4019. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, R. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag New York: New York, NY, USA, 2016. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Andy Liaw, W.H.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. Gplots: Various R Programming Tools For Plotting Data, R package version 3.0. 1; The R Foundation: Vienna, Austria, 2016. [Google Scholar]
- Angeles-Albores, D.L.; Lee, R.Y.; Chan, J.; Sternberg, P.W. Two new functions in the WormBase Enrichment Suite. Micropublication Biol. 2018. [Google Scholar] [CrossRef]
- Hu, M.; UAB, Birmingham, AL, USA. Unpublished work. 2018.
- Greer, E.R.; Perez, C.L.; Van Gilst, M.R.; Lee, B.H.; Ashrafi, K. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab. 2008, 8, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Patterson, G.I.; Padgett, R.W. TGF beta-related pathways. Roles in Caenorhabditis elegans development. Trends Genet. 2000, 16, 27–33. [Google Scholar] [CrossRef]
- Hu, P.J. Dauer. WormBook 2007, 1–19. [Google Scholar] [CrossRef]
- Okumura, N.; Hashimoto, K.; Kitahara, M.; Okuda, H.; Ueda, E.; Watanabe, K.; Nakahara, M.; Sato, T.; Kinoshita, S.; Tourtas, T.; et al. Activation of TGF-beta signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6801. [Google Scholar] [CrossRef]
- Liu, Z.; Li, C.; Kang, N.; Malhi, H.; Shah, V.H.; Maiers, J.L. Transforming growth factor beta (TGFbeta) cross-talk with the unfolded protein response is critical for hepatic stellate cell activation. J. Biol. Chem. 2019, 294, 3137–3151. [Google Scholar] [CrossRef] [Green Version]
- George-Raizen, J.B.; Shockley, K.R.; Trojanowski, N.F.; Lamb, A.L.; Raizen, D.M. Dynamically-expressed prion-like proteins form a cuticle in the pharynx of Caenorhabditis elegans. Biology Open 2014, 3, 1139. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic of the DAF-7/TGFβ pathway in C. elegans. (Adapted from wormbook.org).
Figure 1.
Schematic of the DAF-7/TGFβ pathway in C. elegans. (Adapted from wormbook.org).

Figure 2.
Description of the RNA-seq data. (A) Principle component analysis (PCA) of the sequenced samples. Red dots denote daf-1; daf-3 double mutants, blue dots denote daf-1 single mutants, and green dots denote wild-type N2. (B) The heatmap of the eight samples displayed in the PCA analysis (A). Samples are clustered hierarchically. Values on the x-axis of the color key denote relative z-scores. Count value on the y-axis denote the number of genes belonging to each z value on the x-axis. (C) Venn diagram of the differentially expressed genes that are ≥2-fold difference, q < 0.05 in daf-1 vs. N2, daf-1 vs. daf-1; daf-3, and daf-1; daf-3 vs. N2.
Figure 2.
Description of the RNA-seq data. (A) Principle component analysis (PCA) of the sequenced samples. Red dots denote daf-1; daf-3 double mutants, blue dots denote daf-1 single mutants, and green dots denote wild-type N2. (B) The heatmap of the eight samples displayed in the PCA analysis (A). Samples are clustered hierarchically. Values on the x-axis of the color key denote relative z-scores. Count value on the y-axis denote the number of genes belonging to each z value on the x-axis. (C) Venn diagram of the differentially expressed genes that are ≥2-fold difference, q < 0.05 in daf-1 vs. N2, daf-1 vs. daf-1; daf-3, and daf-1; daf-3 vs. N2.

Figure 3.
General trends of the differentially expressed genes (DEGs). The bar graphs represent the number of up and down regulated genes in daf-1(m40) mutants in (A) All DEGs in the daf-1 vs. N2 dataset. (B) DEGs in the daf-1 vs. N2 dataset that are regulated by daf-1 independent of daf-3. (C) DEGs that are differentially regulated by daf-1 and daf-3. The number above each bar denotes the number of genes.
Figure 3.
General trends of the differentially expressed genes (DEGs). The bar graphs represent the number of up and down regulated genes in daf-1(m40) mutants in (A) All DEGs in the daf-1 vs. N2 dataset. (B) DEGs in the daf-1 vs. N2 dataset that are regulated by daf-1 independent of daf-3. (C) DEGs that are differentially regulated by daf-1 and daf-3. The number above each bar denotes the number of genes.

Figure 4.
qPCR validation of select genes from the RNA-seq dataset. (A) Expression fold change for three Response to Topologically Incorrect Protein Folding genes. (A’) Biological replicate of A. (B) Expression fold change for three Response to Biotic Stimulus genes. (B’) Biological replicate of B. For all qPCR analysis, cDNA was synthesized from 1µg of total RNA. qPCR was performed using 2 µL of cDNA per reaction with gene specific primers and cdc-42 as the endogenous control. Error bars represent standard deviation of technical triplicates.
Figure 4.
qPCR validation of select genes from the RNA-seq dataset. (A) Expression fold change for three Response to Topologically Incorrect Protein Folding genes. (A’) Biological replicate of A. (B) Expression fold change for three Response to Biotic Stimulus genes. (B’) Biological replicate of B. For all qPCR analysis, cDNA was synthesized from 1µg of total RNA. qPCR was performed using 2 µL of cDNA per reaction with gene specific primers and cdc-42 as the endogenous control. Error bars represent standard deviation of technical triplicates.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
GO term enrichment analysis of the genes differentially regulated by daf-1 and daf-3.
| Term | Expected | Observed | Enrichment Fold Change | p Value | q Value |
|---|---|---|---|---|---|
| Genes downregulated in daf-1(m40) mutants | |||||
| Biological Process GO Term | |||||
| Response to biotic stimulus GO:0009607 | 0.1 | 3 | 30 | 3.20 × 10−6 | 0.00041 |
| Genes upregulated in daf-1(m40) mutants | |||||
| Biological Process GO Terms | |||||
| Response to topologically incorrect protein GO:0035966 | 1.3 | 10 | 7.5 | 1.30 × 10−7 | 5.50 × 10−6 |
| IRE1-mediated unfolded protein response GO:0036498 | 0.81 | 4 | 5 | 0.0014 | 0.014 |
| Response to biotic stimulus GO:0009607 | 1.3 | 7 | 5.2 | 7.10 × 10−5 | 0.0015 |
| Molting cycle GO:0042303 | 0.77 | 5 | 6.5 | 0.00014 | 0.0022 |
| Cellular Component GO Terms | |||||
| Collagen trimer GO:0005581 | 1.2 | 30 | 26 | 1.10 × 10−34 | 7.10 × 10−33 |
| Extracellular region GO:0005576 | 3.9 | 16 | 4.1 | 5.50 × 10−7 | 1.70 × 10−5 |
| Extracellular space GO:0005615 | 2 | 7 | 3.4 | 0.0011 | 0.014 |
| Membrane GO:0016020 | 46 | 71 | 1.6 | 9.20 × 10−5 | 0.0017 |
| Intrinsic component of membrane GO:0031224 | 40 | 67 | 1.7 | 2.10 × 10−5 | 0.00054 |
| Molecular Function GO Terms | |||||
| Structural constituent of cuticle GO:0042302 | 1.1 | 32 | 28 | 4.60 × 10−38 | 5.90 × 10−36 |
| Passive transmembrane transporter activity GO:0022803 | 2.5 | 8 | 3.2 | 0.0011 | 0.014 |
| Substrate-specific channel activity GO:0022838 | 2.3 | 8 | 3.5 | 0.00052 | 0.0074 |
| Peptidase activity GO:0008233 | 3.1 | 9 | 2.9 | 0.0014 | 0.014 |
GO enrichment analysis was performed using the Enrichment Analysis tool on wormbase.org. GO terms were manually separated into the major categories. Arrows denote the child of the parent term immediately above.
Table 2.
Genes in each GO term associated with biological processes.
| Genes Upregulated in daf-1(m40) Mutants | |||
|---|---|---|---|
| GO Term: Response to Topologically Incorrect Protein Folding GO: 0035966 | |||
| Gene Name | RNAseq Fold Change | p Value | q Value |
| abu-1 | 6.6291588 | 0.00015 | 0.00184372 |
| * abu-8 | 2.3753599 | 5.00 × 10−5 | 0.00070927 |
| abu-10 | 2.333622 | 5.00 × 10−5 | 0.00070927 |
| * abu-11 | 2.7447639 | 0.0038 | 0.0237475 |
| col-109 | 2.7839511 | 5.00 × 10−5 | 0.00070927 |
| * pqn-74 | 2.3770804 | 5.00 × 10−5 | 0.00070927 |
| fipr-24 | 6.4057217 | 5.00 × 10−5 | 0.00070927 |
| T06E4.8 | 2.1258335 | 0.0002 | 0.00233793 |
| F41E6.11 | 2.7376684 | 5.00 × 10−5 | 0.00070927 |
| clec-67 | 356.70377 | 0.0007 | 0.00656909 |
| GO Term: Response to Biotic Stimulus GO: 0009607 | |||
| Gene name | RNAseq Fold change | p value | q value |
| nlp-30 | 5.5977832 | 5.00 × 10−5 | 0.00070927 |
| fipr-7 | Infinity | 5.00 × 10−5 | 0.00070927 |
| F20G2.5 | 4.2163742 | 5.00 × 10−5 | 0.00070927 |
| *sodh-1 | 2.0632794 | 5.00 × 10−5 | 0.00070927 |
| *clec-61 | 3.9058333 | 5.00 × 10−5 | 0.00070927 |
| irg-1 | 4.981529 | 5.00 × 10−5 | 0.00070927 |
| *math-38 | 8.2638921 | 5.00 × 10−5 | 0.00070927 |
| GO Term: Molting Process GO 0042303 | |||
| Gene name | RNAseq Fold change | p value | q value |
| dpy-13 | 15.645535 | 5.00 × 10−5 | 0.00070927 |
| nas-30 | 2.0286994 | 0.0002 | 0.00233793 |
| rol-6 | 8.4057641 | 0.0003 | 0.00330399 |
| sqt-1 | 12.831644 | 5.00 × 10−5 | 0.00070927 |
| sqt-2 | 5.002505 | 5.00 × 10−5 | 0.00070927 |
| Genes downregulated in daf-1(m40) mutants | |||
| GO Term: Response to Biotic Stimulus GO: 0009607 | |||
| Gene name | RNAseq Fold change | p value | q value |
| ilys-5 | 9.66785229 | 5.00 × 10−5 | 0.00070927 |
| clec-218 | 5.85451214 | 5.00 × 10−5 | 0.00070927 |
| lys-4 | 9.58417075 | 5.00 × 10−5 | 0.00070927 |
* represents genes selected for validation by qPCR.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hu, M.; Crossman, D.; Prasain, J.K.; Miller, M.A.; Serra, R.A. Transcriptomic Profiling of DAF-7/TGFβ Pathway Mutants in C. elegans. Genes 2020, 11, 288. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030288
AMA Style
Hu M, Crossman D, Prasain JK, Miller MA, Serra RA. Transcriptomic Profiling of DAF-7/TGFβ Pathway Mutants in C. elegans. Genes. 2020; 11(3):288. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030288
Chicago/Turabian StyleHu, Muhan, David Crossman, Jeevan K. Prasain, Michael A. Miller, and Rosa A. Serra. 2020. "Transcriptomic Profiling of DAF-7/TGFβ Pathway Mutants in C. elegans" Genes 11, no. 3: 288. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030288
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.