Transcriptome Analysis Identifies LINC00152 as a Biomarker of Early Relapse and Mortality in Acute Lymphoblastic Leukemia


 , , , ,
, , , ,  , add
Show full author list
, add
Show full author list
Abstract
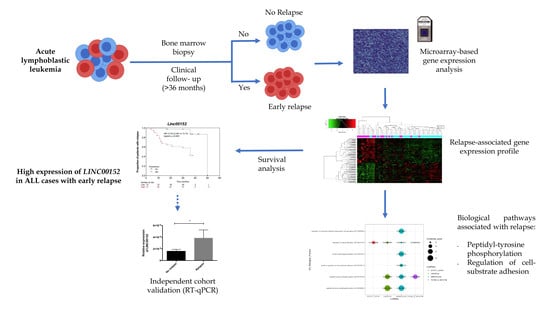
:
1. Introduction
2. Materials and Methods
2.1. Collection of Human Samples
2.2. Total RNA Isolation, Microarray Processing, and Data Normalization
2.3. mRNA and lncRNA Transcriptional Profiling Analysis
2.4. Quantitative Real-Time PCR Validation
2.5. lncRNA–mRNA Co-Expression Analysis and Biological Pathways Analysis
2.6. Validation Cohort
2.7. Statistical Analysis
3. Results
3.1. Clinical and Demographic Features of the Studied Population
3.1.1. Discovery Cohort
3.1.2. Validation Cohort
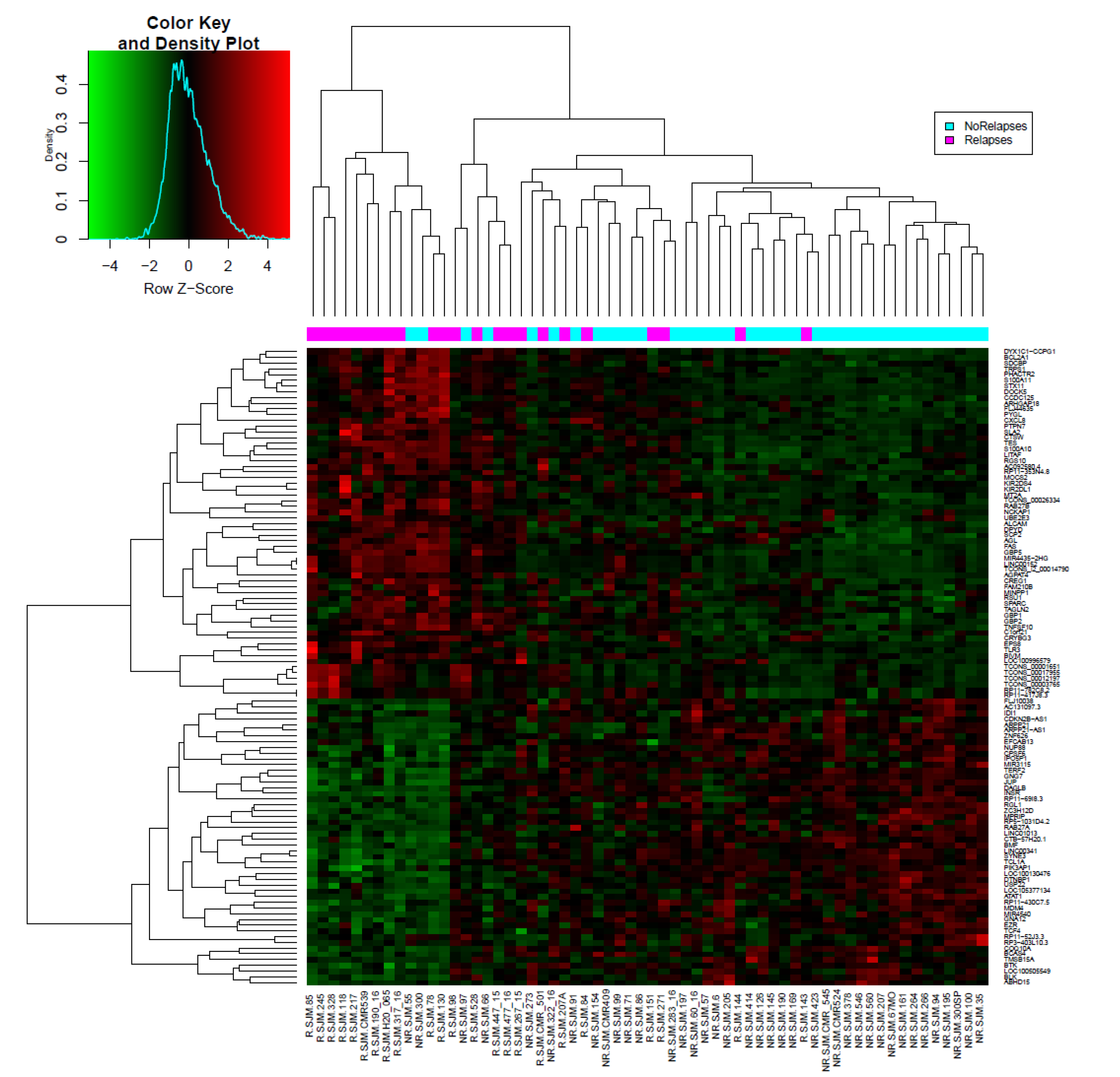
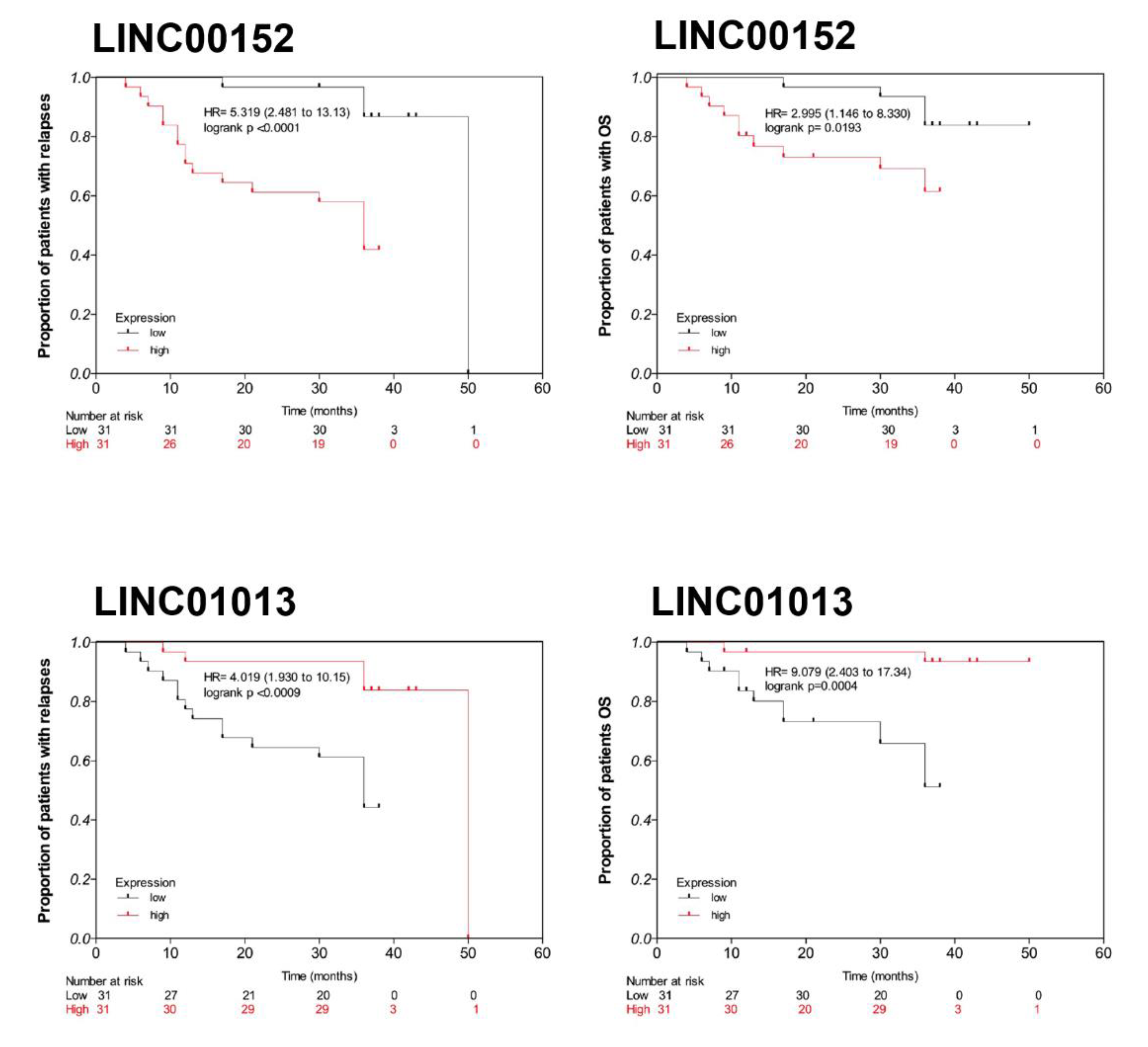
3.2. LINC00152 and LINC01013 Were Abnormally Regulated in Patients with Early Relapse and Early Death
3.3. Up-Regulated lnRNAs in Patients with hyperleukocytosis
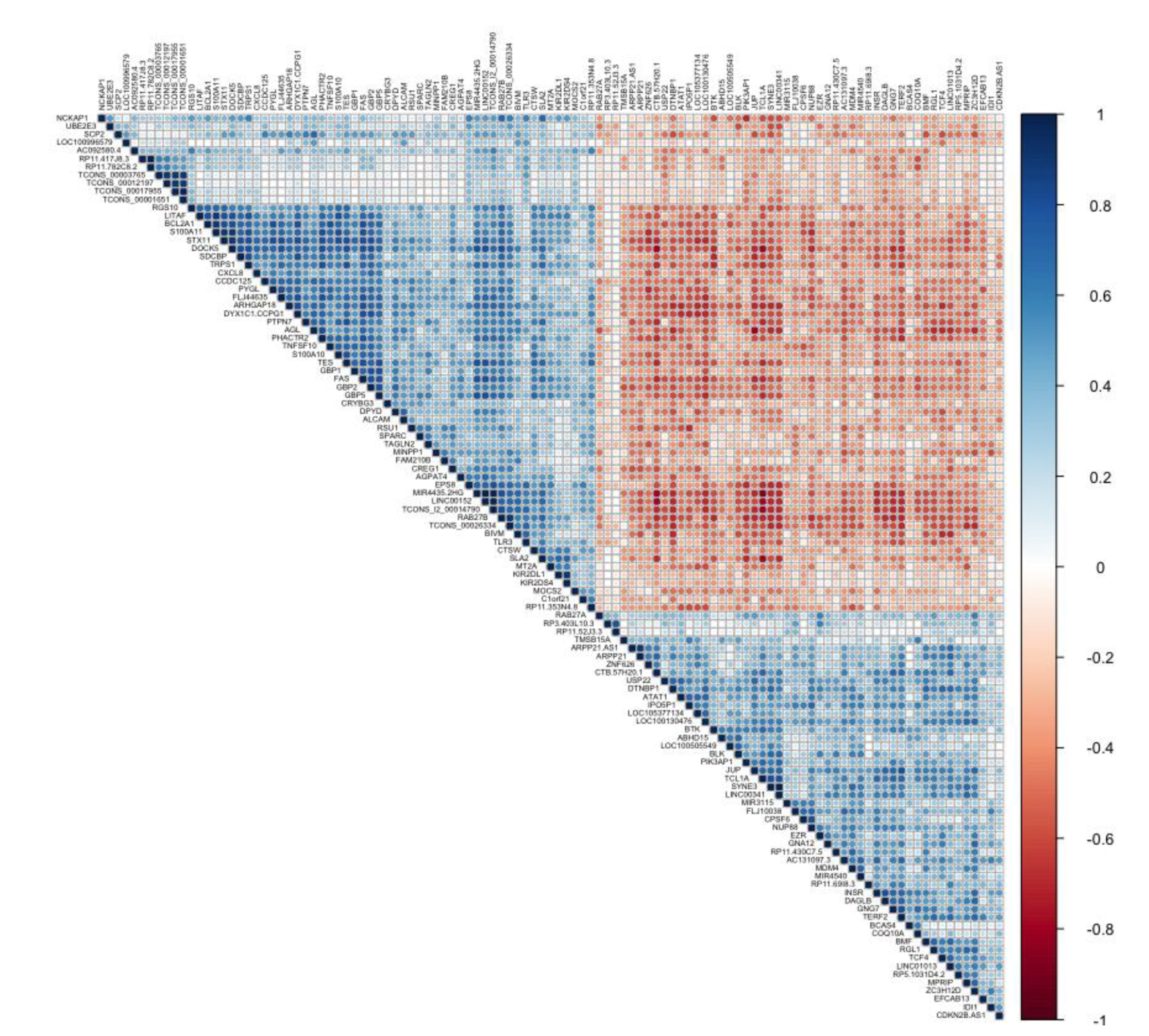
3.4. Co-Expression of lncRNA-Coding Genes and Biological Pathways Analysis
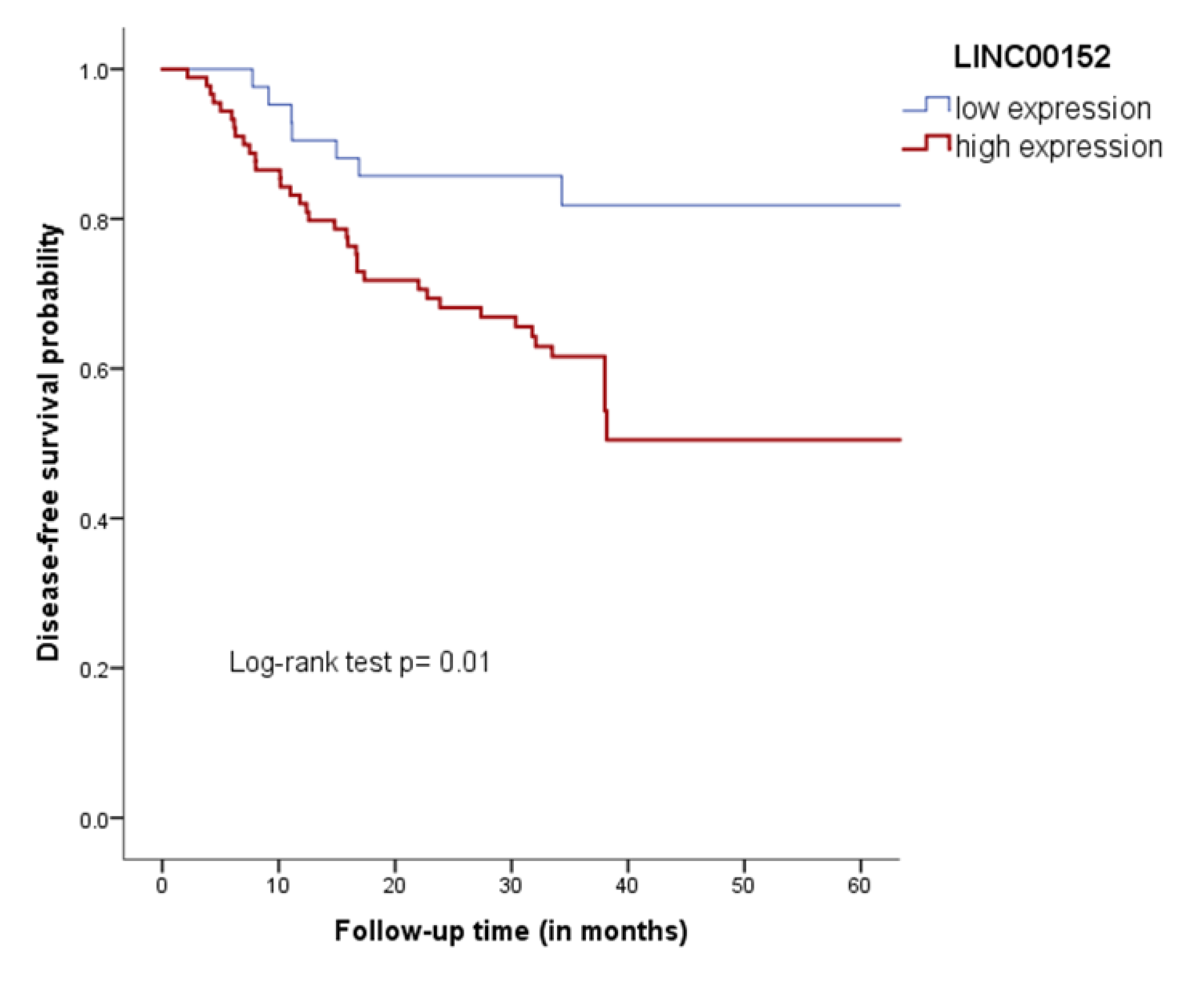
3.5. Clinical Association and Survival Analysis
3.5.1. Discovery Cohort
3.5.2. Validation Cohort
4. Discussion
4.1. LINC00152 and LINC01013 Expression in Acute Lymphoblastic Leukemia
4.2. LncRNAs in Hyperleukocytosis
4.3. LINC00152 and LINC01013 as Potential Biomarkers of Poor Outcomes in Acute Lymphoblastic Leukemia
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barrington-Trimis, J.L.; Cockburn, M.; Metayer, C.; Gauderman, W.J.; Wiemels, J.; McKean-Cowdin, R. Trends in childhood leukemia incidence over two decades from 1992 to 2013. Int. J. Cancer 2017, 140, 1000–1008. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Luna, R.; Velasco-Hidalgo, L.; Zapata-Tarrés, M.; Cárdenas-Cardos, R.; Aguilar-Ortiz, M.R. Current outlook of childhood cancer epidemiology in a middle-income country under a public health insurance program. Pediatr. Hematol. Oncol. 2017, 34, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Linet, M.S.; Brown, L.M.; Mbulaiteye, S.M.; Check, D.; Ostroumova, E.; Landgren, A.; Devesa, S.S. International long-term trends and recent patterns in the incidence of leukemias and lymphomas among children and adolescents ages 0–19 years. Int. J. Cancer 2016, 138, 1862–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Hernández, E.; Jaimes-Reyes, E.Z.; Arellano-Galindo, J.; García-Jiménez, X.; Tiznado-García, H.M.; Dueñas-González, M.T.; Martínez-Villegas, O.; Sánchez-Jara, B.; Bekker-Méndez, V.C.; Ortíz-Torres, M.G.; et al. Survival of Mexican Children with Acute Lymphoblastic Leukaemia under Treatment with the Protocol from the Dana-Farber Cancer Institute 00–01. Biomed. Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Martín-Trejo, J.A.; Núñez-Enríquez, J.C.; Fajardo-Gutiérrez, A.; Medina-Sansón, A.; Flores-Lujano, J.; Jiménez-Hernández, E.; Amador-Sanchez, R.; Peñaloza-González, J.G.; Álvarez-Rodríguez, F.J.; Bolea-Murga, V.; et al. Early mortality in children with acute lymphoblastic leukemia in a developing country: The role of malnutrition at diagnosis. A multicenter cohort MIGICCL study. Leuk. Lymphoma. 2017, 58, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Han, B.W.; Chen, Z.H.; Lin, K.Y.; Zeng, C.W.; Li, X.J.; Li, J.H.; Luo, X.Q.; Chen, Y.Q. A distinct set of long non-coding RNAs in childhood MLL-rearranged acute lymphoblastic leukemia: Biology and epigenetic target. Hum. Mol. Genet. 2014, 23, 3278–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, T.R.; Rodriguez-Malave, N.I.; Waters, E.V.; Yan, W.; Casero, D.; Basso, G.; Pigazzi, M.; Rao, D.S. LncRNA Expression Discriminates Karyotype and Predicts Survival in B-Lymphoblastic Leukemia. Mol. Cancer Res. 2015, 13, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Xiao, P.F.; Tao, Y.F.; Hu, S.Y.; Lu, J.; Zhao, W.L.; Li, Z.H.; Wang, N.N.; Wang, J.; Feng, X.; et al. Microarray profiling of bone marrow long non-coding RNA expression in Chinese pediatric acute myeloid leukemia patients. Oncol. Rep. 2016, 35, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Dominguez, J.R.; Lodish, H.F. Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood 2017, 130, 1965–1975. [Google Scholar] [CrossRef]
- Lammens, T.; Durinck, K.; Wallaert, A.; Speleman, F.; Van Vlierberghe, P. Long non-coding RNAs in leukemia: Biology and clinical impact. Curr. Opin. Hematol. 2017, 24, 353–358. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, H.G.; Lu, C. A novel long non-coding RNA T-ALL-R-LncR1 knockdown and Par-4 cooperate to induce cellular apoptosis in T-cell acute lymphoblastic leukemia cells. Leuk. Lymphoma 2014, 55, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Ghazavi, F.; De Moerloose, B.; Van Loocke, W.; Wallaert, A.; Helsmoortel, H.H.; Ferster, A.; Bakkus, M.; Plat, G.; Delabesse, E.; Uyttebroeck, A.; et al. Unique long non-coding RNA expression signature in ETV6/RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Oncotarget 2016, 7, 73769–73780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallaert, A.; Durinck, K.; Van Loocke, W.; Van de Walle, I.; Matthijssens, F.; Volders, P.J.; Avila-Cobos, F.; Rombaut, D.; Rondou, P.; Mestdagh, P.; et al. Long noncoding RNA signatures define oncogenic subtypes in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Gioia, R.; Drouin, S.; Ouimet, M.; Caron, M.; St-Onge, P.; Richer, C.; Sinnett, D. LncRNAs downregulated in childhood acute lymphoblastic leukemia modulate apoptosis, cell migration, and DNA damage response. Oncotarget 2017, 8, 80645–80650. [Google Scholar] [CrossRef] [Green Version]
- Ngoc, P.C.T.; Tan, S.H.; Tan, T.K.; Chan, M.M.; Li, Z.; Yeoh, A.E.J.; Tenen, D.G.; Sanda, T. Identification of novel lncRNAs regulated by the TAL1 complex in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 2138–2151. [Google Scholar] [CrossRef]
- James, A.R.; Schroeder, M.P.; Neumann, M.; Bastian, L.; Eckert, C.; Gökbuget, N.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Schwartz, S.; et al. Long non-coding RNAs defining major subtypes of B cell precursor acute lymphoblastic leukemia. J. Hematol. Oncol. 2019, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Rosa, R.; Villegas-Ruíz, V.; Caballero-Palacios, M.C.; Pérez-López, E.I.; Murata, C.; Zapata-Tarres, M.; Cárdenas-Cardos, R.; Paredes-Aguilera, R.; Rivera-Luna, R.; Juárez-Méndez, A.S. Expression of ZNF695 Transcript Variants in Childhood B-Cell Acute Lymphoblastic Leukemia. Genes 2019, 10, 716. [Google Scholar] [CrossRef] [Green Version]
- Núñez-Enríquez, J.C.; Bárcenas-López, D.A.; Hidalgo-Miranda, A.; Jiménez-Hernández, E.; Bekker-Méndez, V.C.; Flores-Lujano, J.; Solís-Labastida, K.A.; Martínez-Morales, G.B.; Sánchez-Muñoz, F.; Espinoza-Hernández, L.E.; et al. Gene Expression Profiling of Acute Lymphoblastic Leukaemia in Children with Very Early Relapse. Arch. Med. Res. 2016, 47, 644–655. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Tehrani, S.S.; Karimian, A.; Parsian, H.; Majidinia, M.; Yousefi, B. Multiple Functions of Long Non-Coding RNAs in Oxidative Stress, DNA Damage Response and Cancer Progression. J. Cell. Biochem. 2018, 119, 223–236. [Google Scholar] [CrossRef]
- Cao, W.J.; Wu, H.L.; He, B.S.; Zhang, Y.S.; Zhang, Z.Y. Analysis of long non-coding RNA expression profiles in gastric cancer. World J. Gastroenterol. 2013, 19, 3658–3664. [Google Scholar] [CrossRef] [PubMed]
- Morlando, M.; Ballarino, M.; Fatica, A. Long Non-Coding RNAs: New Players in Hematopoiesis and Leukaemia. Front. Med. 2015, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Raulefs, S.; Bruns, P.; Afonso-Grunz, F.; Plötner, A.; Thermann, R.; Jäger, C.; Schlitter, A.M.; Kong, B.; Regel, I.; et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol. Cancer 2015, 14, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Dong, Z.; Shi, Y.; Liu, S.; Liang, J.; Guo, Y.; Guo, X.; Shen, S.; Wang, G. Methylation-mediated downregulation of long noncoding RNA LOC100130476 in gastric cardia adenocarcinoma. Clin. Exp. Metastasis 2016, 33, 497–508. [Google Scholar] [CrossRef]
- Guo, W.; Dong, Z.; Shi, Y.; Liu, S.; Liang, J.; Guo, Y.; Guo, X.; Shen, S.; Shan, B. Aberrant methylation-mediated downregulation of long noncoding RNA LOC100130476 correlates with malignant progression of esophageal squamous cell carcinoma. Dig. Liver Dis. 2016, 48, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Ke, D.; Li, H.; Zhang, Y.; An, Y.; Fu, H.; Fang, X.; Zheng, X. The combination of circulating long noncoding RNAs AK001058, INHBA-AS1, MIR4435–2HG, and CEBPA-AS1 fragments in plasma serve as diagnostic markers for gastric cancer. Oncotarget 2017, 8, 21516–21525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.L.; Wang, J.; He, W.; Zhao, P.; Wu, W.Q. Down-regulation of lncRNA Linc00152 suppressed cell viability, invasion, migration, and epithelial to mesenchymal transition, and reversed chemo-resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3074–3084. [Google Scholar] [CrossRef]
- Ma, P.; Wang, H.; Sun, J.; Liu, H.; Zheng, C.; Zhou, X.; Lu, Z. LINC00152 promotes cell cycle progression in hepatocellular carcinoma via miR-193a/b-3p/CCND1 axis. Cell Cycle 2018, 17, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Chung, I.H.; Lu, P.H.; Lin, Y.H.; Tsai, M.M.; Lin, Y.W.; Yeh, C.T.; Lin, K.H. The long non-coding RNA LINC01013 enhances invasion of human anaplastic large-cell lymphoma. Sci. Rep. 2017, 7, 295. [Google Scholar] [CrossRef]
- Bian, Z.; Zhang, J.; Li, M.; Feng, Y.; Yao, S.; Song, M.; Qi, X.; Fei, B.; Yin, Y.; Hua, D.; et al. Long non-coding RNA LINC00152 promotes cell proliferation, metastasis, and confers 5-FU resistance in colorectal cancer by inhibiting miR-139–5p. Oncogenesis 2017, 6, 395. [Google Scholar] [CrossRef]
- Yue, B.; Cai, D.; Liu, C.; Fang, C.; Yan, D. Linc00152 Functions as a Competing Endogenous RNA to Confer Oxaliplatin Resistance and Holds Prognostic Values in Colon Cancer. Mol. Ther. 2016, 24, 2064–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, R.J.; Maglieri, G.; Gutschner, T.; Diederichs, S.; Lund, A.H.; Nielsen, B.S.; Holmstrøm, K. Evaluation of fluorescence in situ hybridization techniques to study long non-coding RNA expression in cultured cells. Nucleic Acids Res. 2018, 46, e4. [Google Scholar] [CrossRef] [PubMed]
- Nötzold, L.; Frank, L.; Gandhi, M.; Polycarpou-Schwarz, M.; Groß, M.; Gunkel, M.; Beil, N.; Erfle, H.; Harder, N.; Rohr, K.; et al. The long non-coding RNA LINC00152 is essential for cell cycle progression through mitosis in HeLa cells. Sci. Rep. 2017, 7, 2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.M.; Huang, M.D.; Sun, D.P.; Kong, R.; Xu, T.P.; Xia, R.; Zhang, E.B.; Shu, Y.Q. Long intergenic non-coding RNA 00152 promotes tumor cell cycle progression by binding to EZH2 and repressing p15 and p21 in gastric cancer. Oncotarget 2016, 7, 9773–9787. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yidayitula, Y.; Zhao, H.; Luo, Y.; Ma, X.; Xu, M. LncRNA LINC00152 promoted glioblastoma progression through targeting the miR-107 expression. Environ. Sci. Pollut. Res. Int. 2018, 25, 17674–17681. [Google Scholar] [CrossRef]
- Reon, B.J.; Takao-Real-Karia, B.; Kiran, M.; Dutta, A. LINC00152 Promotes Invasion through a 3′-Hairpin Structure and Associates with Prognosis in Glioblastoma. Mol. Cancer Res. 2018, 16, 1470–1482. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Sui, J.; Xu, S.Y.; Liang, G.Y.; Pu, Y.P.; Yin, L.H. Comprehensive analysis of a novel four-lncRNA signature as a prognostic biomarker for human gastric cancer. Oncotarget 2017, 8, 75007–75024. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Chen, L.; Huang, J.; Wang, X.; Ma, S.; Cui, F.; Luo, L.; Ling, L.; Luo, K.; Zheng, G. The lncRNA MIR4435–2HG promotes lung cancer progression by activating β-catenin signalling. J. Mol. Med. 2018, 96, 753–764. [Google Scholar] [CrossRef]
- Li, T.; Xu, C.; Cai, B.; Zhang, M.; Gao, F.; Gan, J. Expression and clinicopathological significance of the lncRNA HOXA11-AS in colorectal cancer. Oncol. Lett. 2016, 12, 4155–4160. [Google Scholar] [CrossRef] [Green Version]
- Barwe, S.P.; Quagliano, A.; Gopalakrishnapillai, A. Eviction from the sanctuary: Development of targeted therapy against cell adhesion molecules in acute lymphoblastic leukaemia. Semin. Oncol. 2017, 44, 101–112. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Z.; Luo, W.; Liu, J.; Lou, Y.; Xia, S. NET1 Enhances Proliferation and Chemoresistance in Acute Lymphoblastic Leukaemia Cells. Oncol. Res. 2019, 27, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Wang, J.; Lu, T.; Ma, D.; Wei, D.; Guo, Y.; Cheng, B.; Wang, W.; Fang, Q. Overexpression of heme oxygenase-1 in microenvironment mediates vincristine resistance of B-cell acute lymphoblastic leukaemia by promoting vascular endothelial growth factor secretion. J. Cell. Biochem. 2019, 120, 17791–17810. [Google Scholar] [CrossRef] [PubMed]
- Misawa, A.; Takayama, K.; Urano, T.; Inoue, S. Androgen-induced Long Noncoding RNA (lncRNA) SOCS2-AS1 Promotes Cell Growth and Inhibits Apoptosis in Prostate Cancer Cells. J. Biol. Chem. 2016, 291, 17861–17880. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Liang, M.; Yao, W.; Liu, J.; Niu, Q.; Chen, J.; Liu, Z.; Li, M.; Shi, B.; Pan, J. Functional role of lncRNA LOC101927497 in N-methyl-N′-nitro-N-nitrosoguanidine-induced malignantly transformed human gastric epithelial cells. Life Sci. 2018, 193, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, Y.; Huarte, M. Long non-coding RNAs: Challenges for diagnosis and therapies. Nucleic Acid Ther. 2013, 23, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wei, X.; Liu, Z.; Cao, D.; Tang, Y.; Zou, Z.; Zhou, C.; Lu, Y. Long noncoding RNA CYTOR in cancer: A TCGA data review. Clin. Chim. Acta 2018, 483, 227–233. [Google Scholar] [CrossRef]
- Zhang, X.; Tao, W. Long Noncoding RNA LINC00152 Facilitates the Leukemogenesis of Acute Myeloid Leukemia by Promoting CDK9 through miR-193a. DNA Cell Biol. 2019, 38, 236–242. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | ALL Children n = 62 | p * | |
|---|---|---|---|
| Early Relapse | |||
| No | Yes | ||
| n = 39 | n = 23 | ||
| Gender | n (%) | n (%) | 0.35 |
| Female | 19 (48.7) | 9 (39.1) | |
| Male | 20 (51.3) | 14 (60.9) | |
| Age group (years) | 0.0001 | ||
| 1–9.99 | 29 (74.4) | 8 (34.8) | |
| ≥10 | 10 (25.6) | 15 (65.2) | |
| Age at diagnosis (months) | 0.002 ** | ||
| Median (min–max) | 73 (14–191) | 140 (18–208) | |
| WBC count at diagnosis (×109/L) | 0.75 | ||
| <10 | 13 (33.3) | 5 (21.7) | |
| 10–49.99 | 12 (30.8) | 9 (39.1) | |
| 50–99.99 | 5 (12.8) | 4 (17.4) | |
| ≥100 | 9 (23.1) | 5 (21.7) | |
| BM blast (%) at diagnosis | 0.75 | ||
| <90 | 7 (17.9) | 5 (21.7) | |
| ≥90 | 32 (82.1) | 18 (78.3) | |
| Gene rearrangement | 0.26 | ||
| ETV6-RUNX1 | 7 (17.9) | 5 (21.7) | |
| TCF3-PBX1 | 4 (10.3) | 0 (0) | |
| BCR-ABL1 | 5 (12.8) | 2 (10) | |
| Non-detected | 23 (59.0) | 16 (68.3) | |
| NCI risk classification | 0.004 | ||
| Standard | 20 (51.3) | 4 (14.3) | |
| High | 19 (48.7) | 24 (85.7) | |
| Relapse site | - | ||
| Isolated BM | - | 17 (74) | |
| Isolated CNS | - | 3 (13) | |
| BM and CNS | - | 2 (8.7) | |
| BM and testicular | - | 1 (4.3) | |
| Chemotherapy regimen | |||
| DFCI (reference) | 10 (25.6) | 2 (8.7) | - |
| BFM-95 | 4 (10.3) | 2 (8.7) | 0.43 |
| St Jude Total XV | 16 (41.0) | 8 (34.8) | 0.3 |
| St Jude Total XIIIB | 9 (23.1) | 11 (47.8) | 0.04 |
| Death | <0.0001 | ||
| Yes | 0 (0) | 15 (65.2) | |
| No | 39 (100) | 8 (34.8) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bárcenas-López, D.A.; Núñez-Enríquez, J.C.; Hidalgo-Miranda, A.; Beltrán-Anaya, F.O.; May-Hau, D.I.; Jiménez-Hernández, E.; Bekker-Méndez, V.C.; Flores-Lujano, J.; Medina-Sansón, A.; Tamez-Gómez, E.L.; et al. Transcriptome Analysis Identifies LINC00152 as a Biomarker of Early Relapse and Mortality in Acute Lymphoblastic Leukemia. Genes 2020, 11, 302. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030302
Bárcenas-López DA, Núñez-Enríquez JC, Hidalgo-Miranda A, Beltrán-Anaya FO, May-Hau DI, Jiménez-Hernández E, Bekker-Méndez VC, Flores-Lujano J, Medina-Sansón A, Tamez-Gómez EL, et al. Transcriptome Analysis Identifies LINC00152 as a Biomarker of Early Relapse and Mortality in Acute Lymphoblastic Leukemia. Genes. 2020; 11(3):302. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030302
Chicago/Turabian StyleBárcenas-López, Diego Alberto, Juan Carlos Núñez-Enríquez, Alfredo Hidalgo-Miranda, Fredy Omar Beltrán-Anaya, Didier Ismael May-Hau, Elva Jiménez-Hernández, Vilma Carolina Bekker-Méndez, Janet Flores-Lujano, Aurora Medina-Sansón, Edna Liliana Tamez-Gómez, and et al. 2020. "Transcriptome Analysis Identifies LINC00152 as a Biomarker of Early Relapse and Mortality in Acute Lymphoblastic Leukemia" Genes 11, no. 3: 302. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030302