Genome-Wide Analysis and Function Prediction of Long Noncoding RNAs in Sheep Pituitary Gland Associated with Sexual Maturation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Samples Collection
2.2. Plasma Hormone Assay
2.3. RNA Extraction and Strand-Specific cDNA Library Construction
2.4. Data Analysis and Transcripts Assembly
2.5. New lncRNA Identification and Classification
2.6. Differential Expression Analysis of mRNA and lncRNA
2.7. Functional Enrichment Analysis of DEGs
2.8. Targeted Gene Prediction Analysis of lncRNA
2.9. lncRNA-mRNA Co-Expressed Network Construction
2.10. Sheep Pituitary Cell Isolation and Cell Transfection
2.11. Real-time PCR Analysis
2.12. Statistical Analysis
3. Results
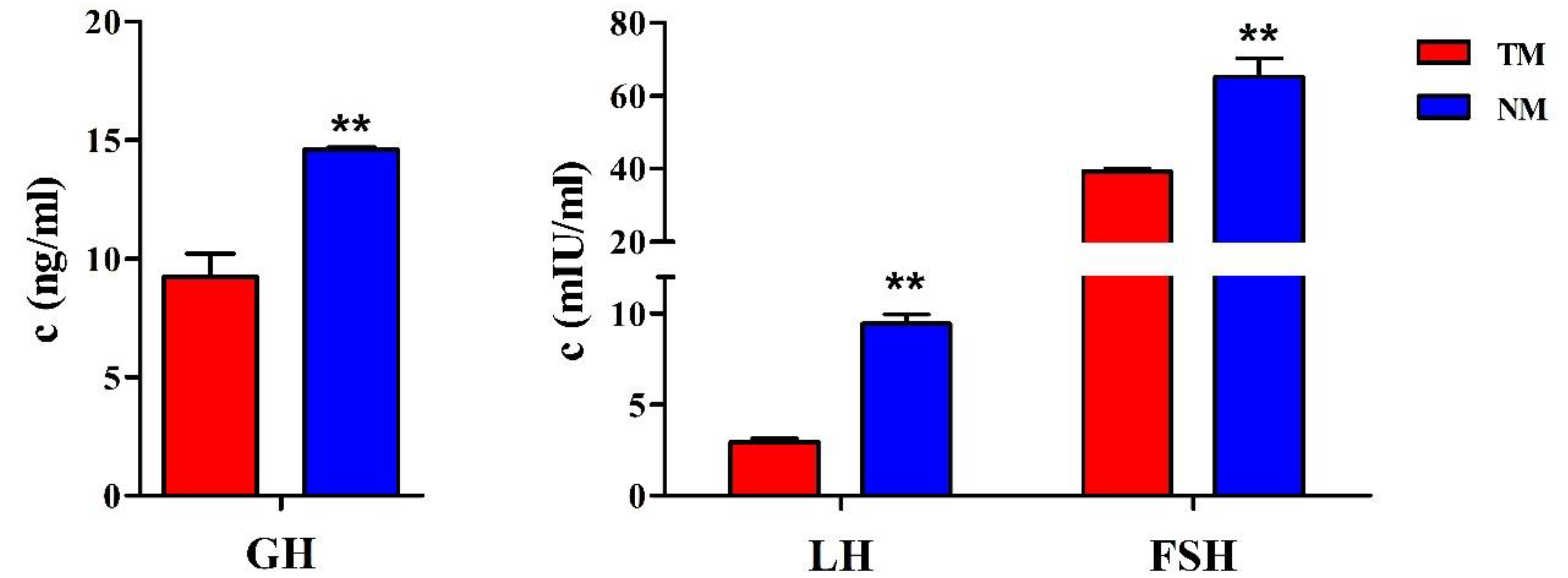
3.1. The Hormone Levels in Immature and Mature Sheep
3.2. Overview of Sequencing in Sheep Pituitary Tissues
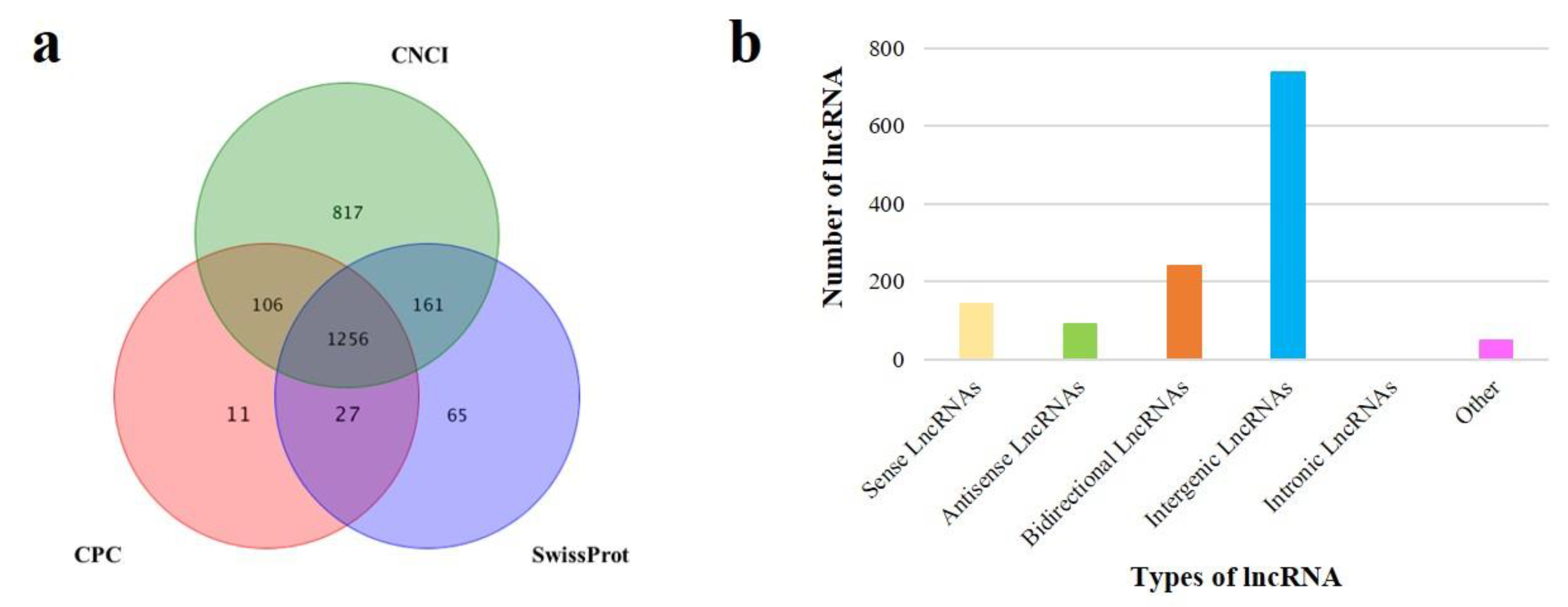
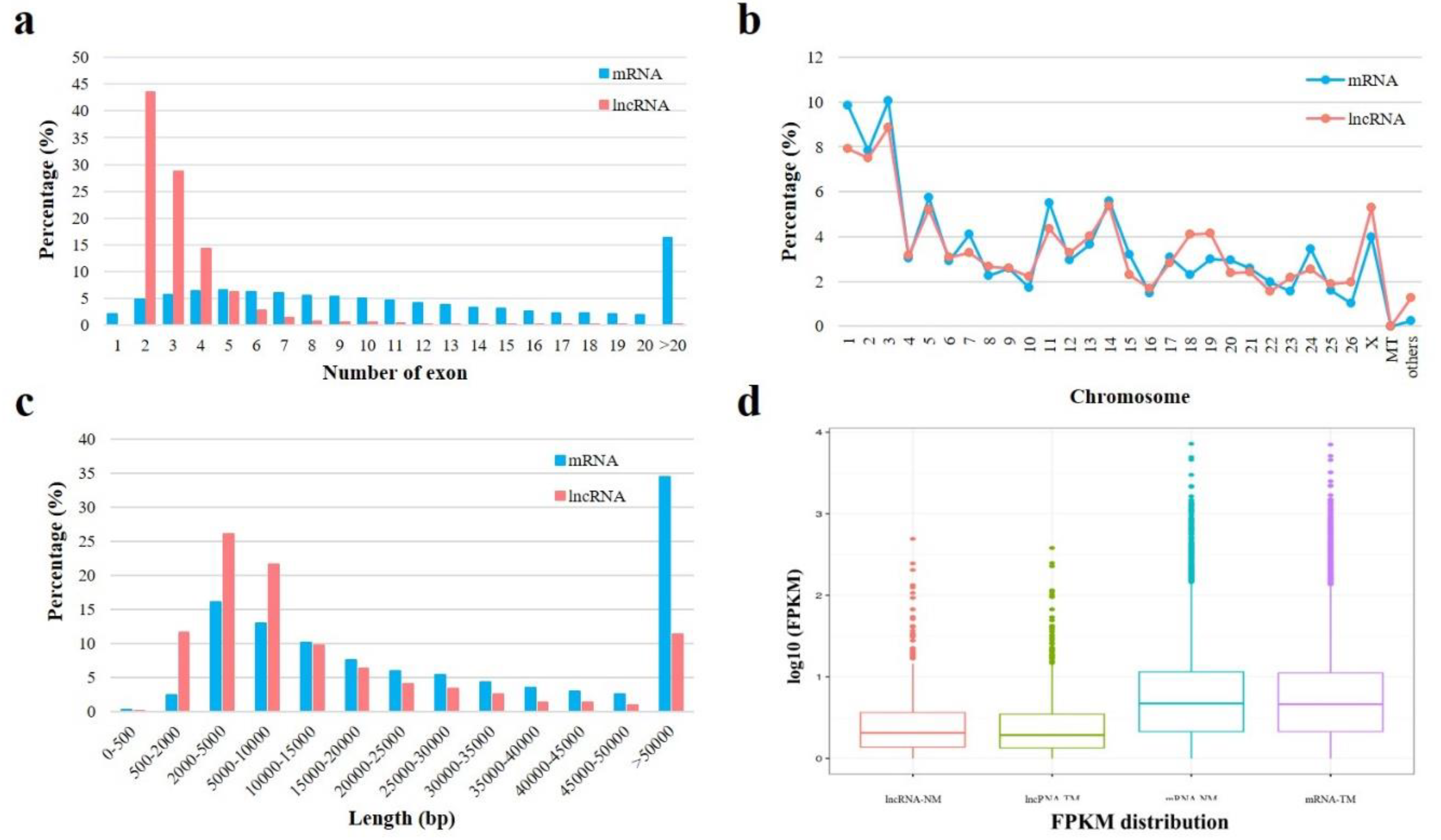
3.3. Identification of lncRNAs and mRNAs in Sheep Pituitary
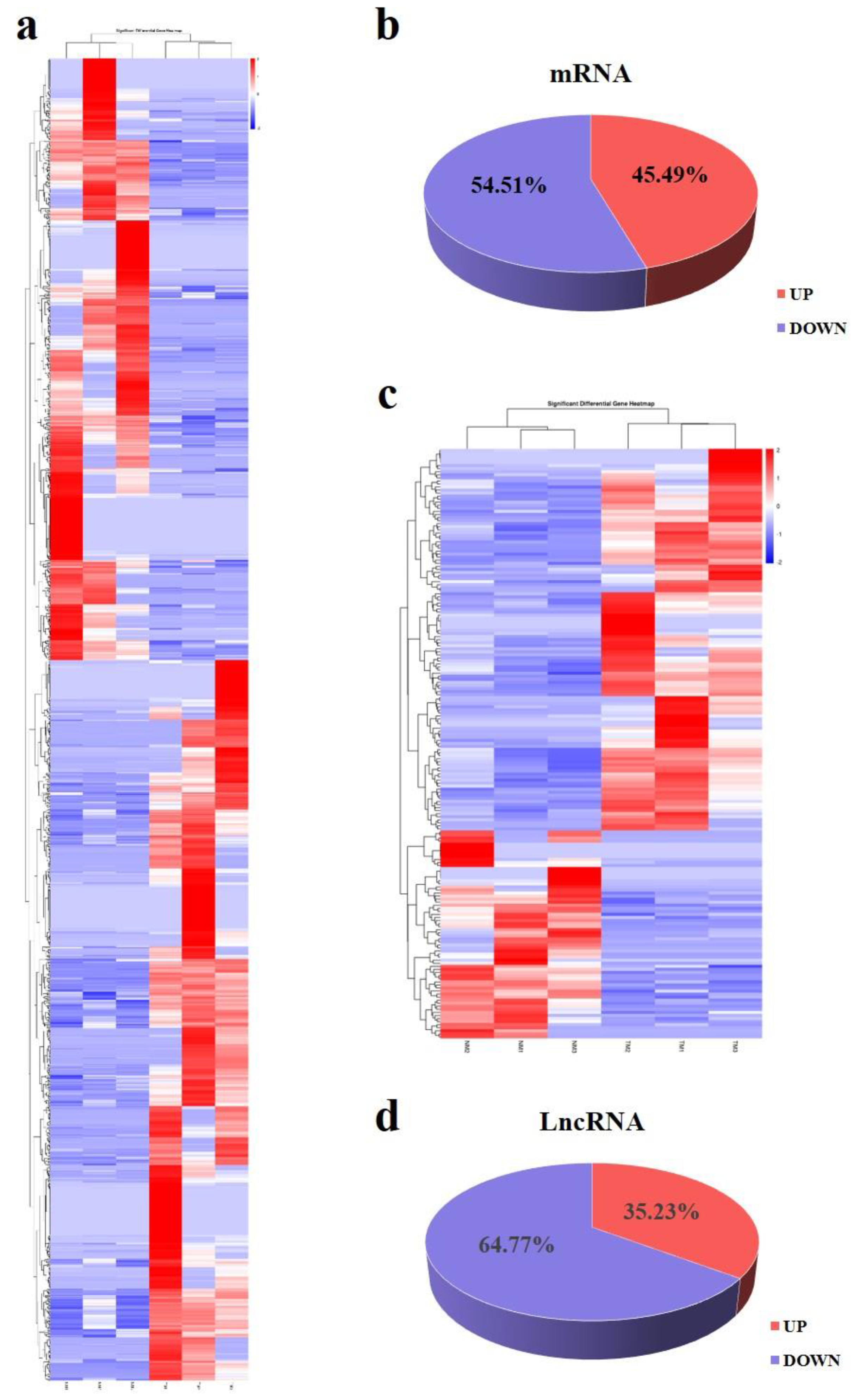
3.4. Differentially Expressed lncRNAs and mRNAs in Immature and Mature Sheep Pituitary
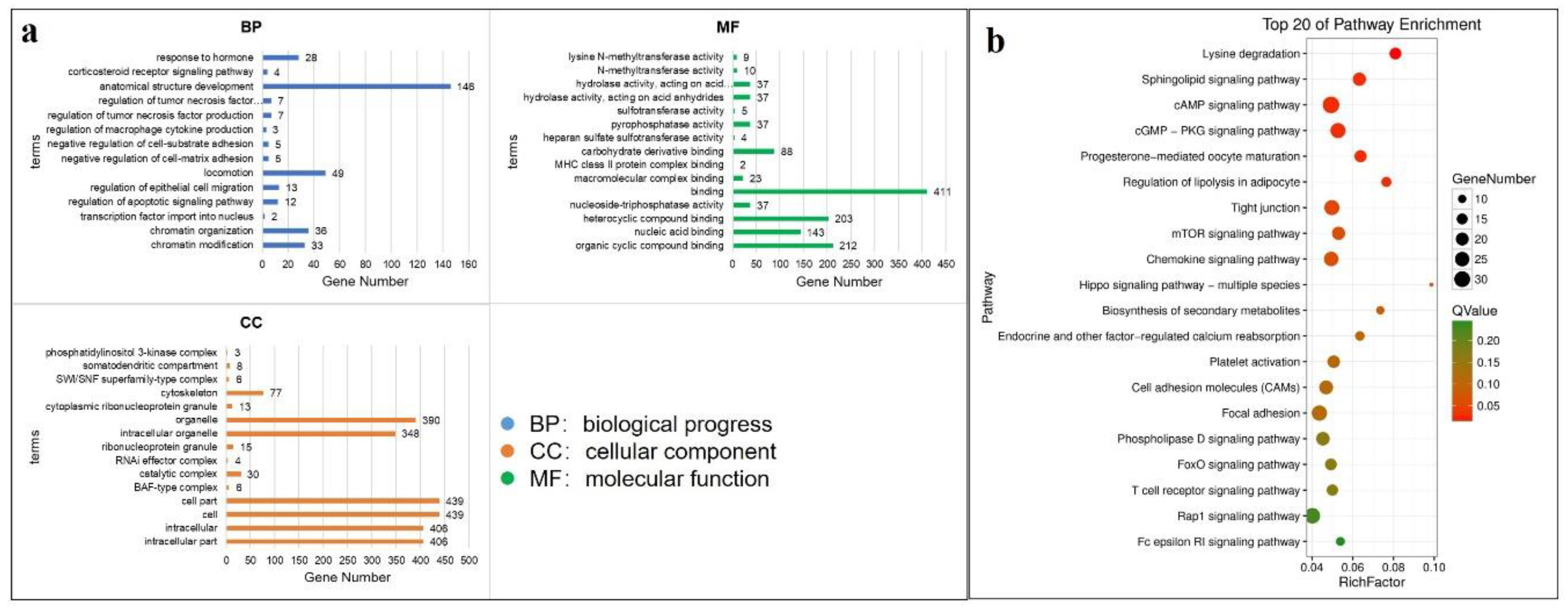
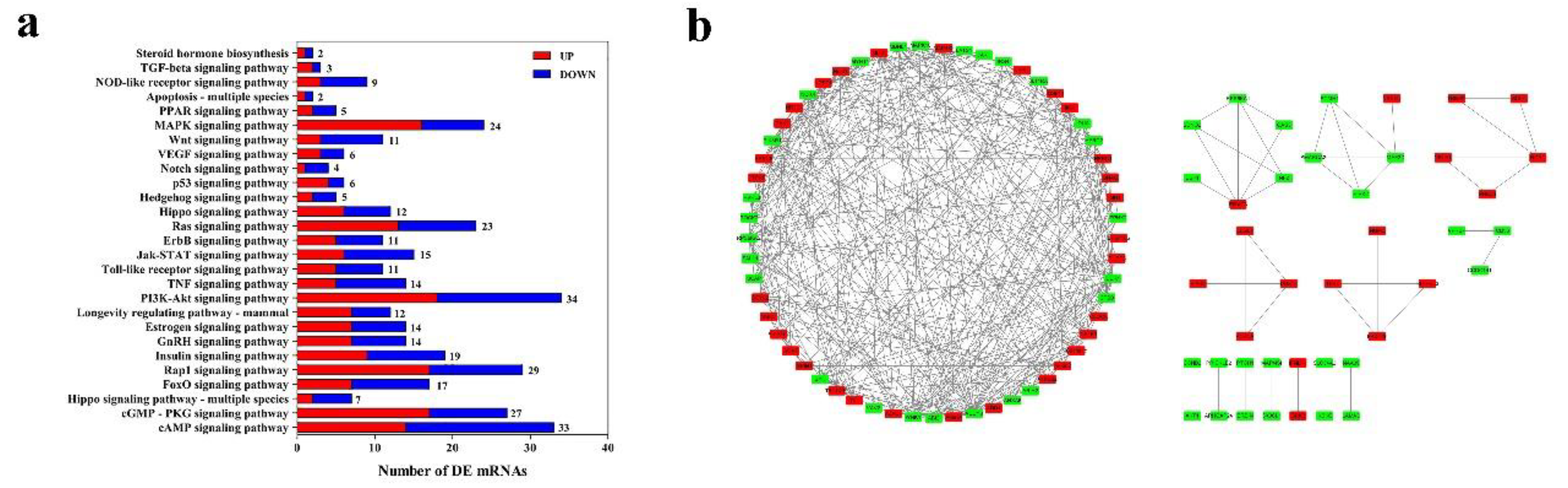
3.5. Functional Enrichment Analysis and Crucial Gene Filter of DE mRNAs
3.6. Interaction Analysis of Candidate lncRNAs and Their Targeted mRNAs
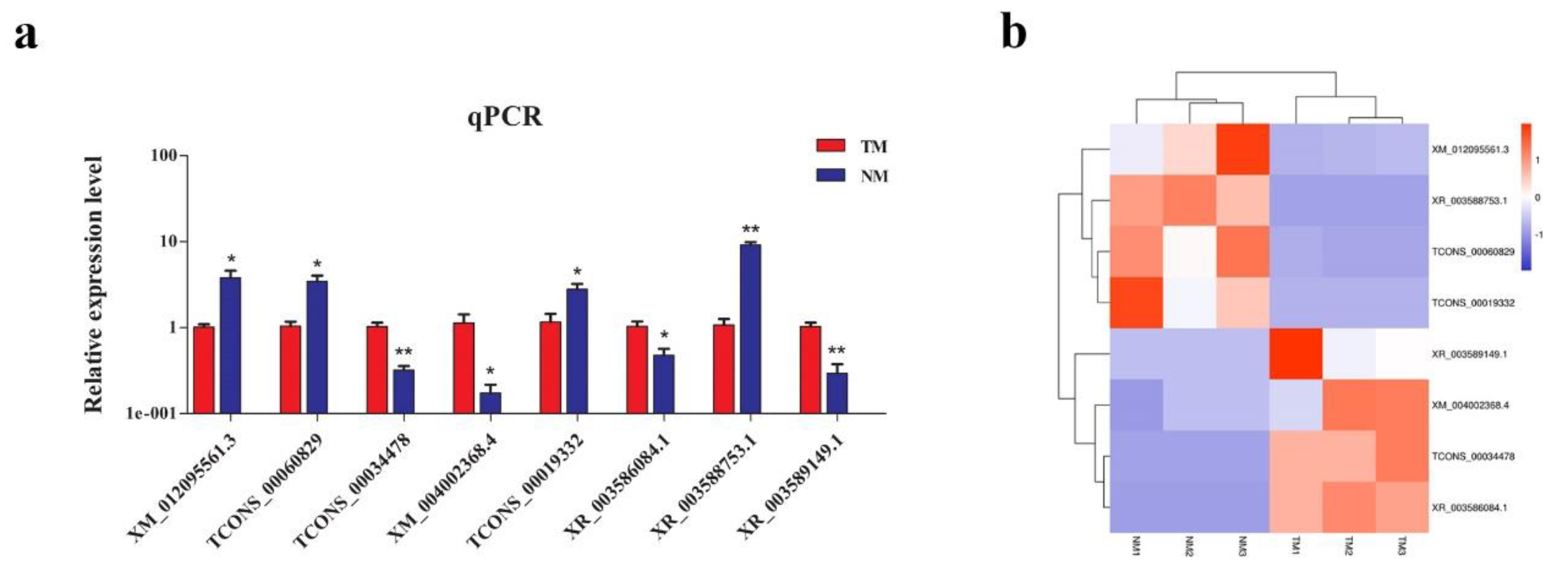
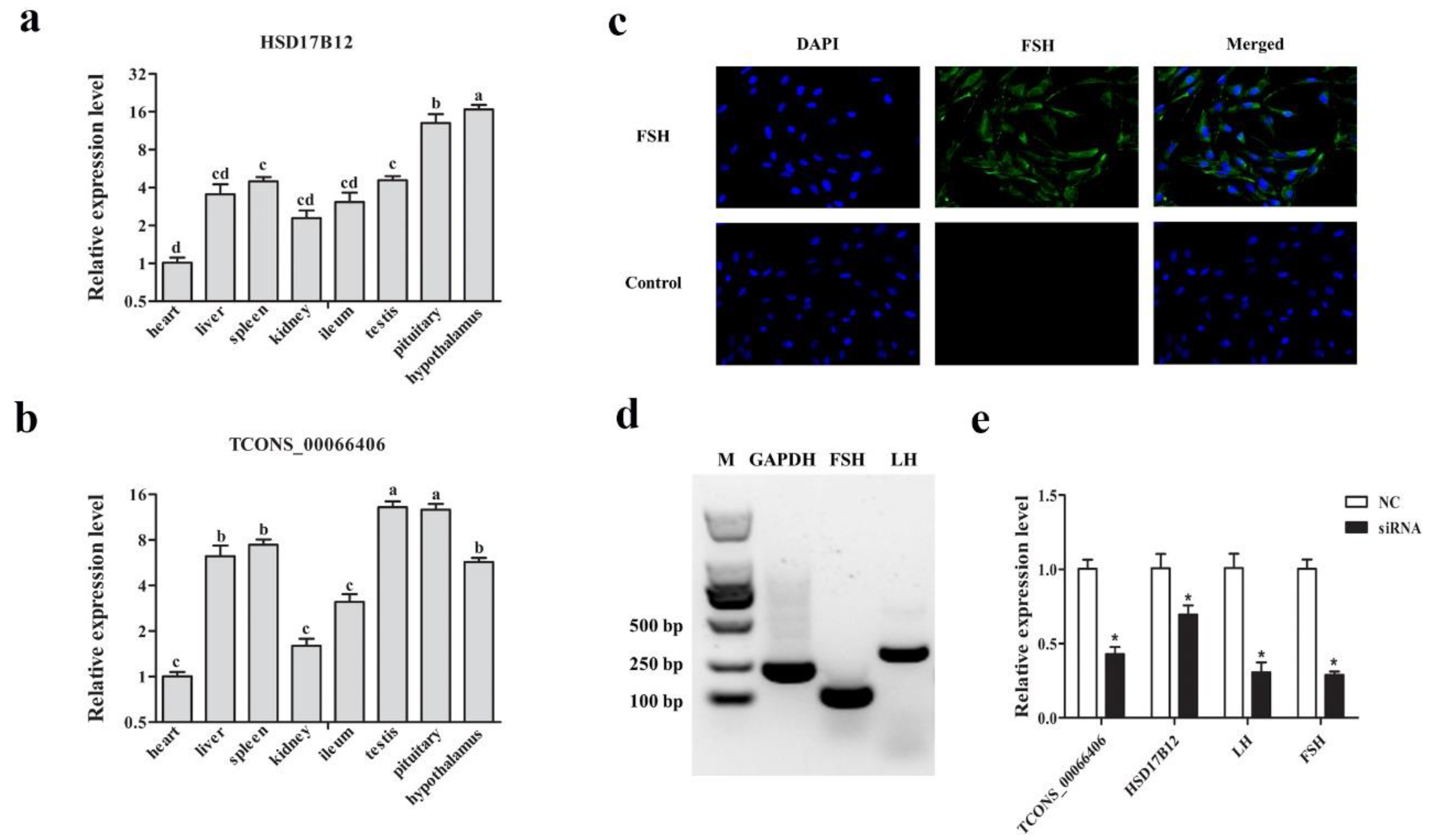
3.7. Verification of Targeting Relationship between lncRNA TCONS_00066406 and Its Targeted Gene HSD17B12 in Sheep Pituitary
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Devaux, Y.; Zangrando, J.; Schroen, B.; Creemers, E.E.; Pedrazzini, T.; Chang, C.P.; Dorn, G.W.; Thum, T.; Heymans, S.; Network, C. Long noncoding rnas in cardiac development and ageing. Nat. Rev. Cardiol. 2015, 12, 415–425. [Google Scholar] [PubMed]
- Taylor, D.H.; Chu, E.T.; Spektor, R.; Soloway, P.D. Long non-coding rna regulation of reproduction and development. Mol. Reprod. Dev. 2015, 82, 932–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.H. Long non-coding rnas and complex diseases: From experimental results to computational models. Brief. Bioinform. 2017, 18, 558–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Froberg, J.E.; Lee, J.T. Long noncoding rnas: Fresh perspectives into the rna world. Trends Biochem. Sci. 2014, 39, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding rnas: Functional surprises from the rna world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Fritah, S.; Niclou, S.P.; Azuaje, F. Databases for lncrnas: A comparative evaluation of emerging tools. Rna Publ. Rna Soc. 2014, 20, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Pieter-Jan, V.; Kenneth, V.; Gerben, M.; Klaas, V.; Lennart, M.; Jo, V.; Pieter, M. An update on lncipedia: A database for annotated human lncrna sequences. Nucleic Acids Res. 2015, 43, 174–180. [Google Scholar]
- Pieter-Jan, V.; Kenny, H.; Xiaowei, W.; Björn, M.; Lennart, M.; Kris, G.; Jo, V.; Pieter, M. Lncipedia: A database for annotated human lncrna transcript sequences and structures. Nucleic Acids Res. 2013, 41, 246–251. [Google Scholar]
- Cheng, Q.X.; Thomson, D.W.; Maag, J.L.V.; Nenad, B.; Bethany, S.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. Lncrnadb v2.0: Expanding the reference database for functional long noncoding rnas. Nucleic Acids Res. 2015, 43, 168–173. [Google Scholar]
- Dechao, B.; Kuntao, Y.; Silong, S.; Chaoyong, X.; Geir, S.; Ruoyu, M.; Hui, X.; Qi, L.; Haitao, L.; Guoguang, Z. Noncode v3.0: Integrative annotation of long noncoding rnas. Nucleic Acids Res. 2012, 40, D210–D215. [Google Scholar]
- Geng, C.; Ziyun, W.; Dongqing, W.; Chengxiang, Q.; Mingxi, L.; Xing, C.; Qipeng, Z.; Guiying, Y.; Qinghua, C. Lncrnadisease: A database for long-non-coding rna-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar]
- Rizzoti, K. Genetic regulation of murine pituitary development. J. Mol. Endocrinol. 2015, 54, R55–R73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Hoover, A.R.; Dozmorov, I.; Raj, P.; Khan, S.; Molina, E.; Chang, T.C.; de la Morena, M.T.; Cleaver, O.B.; Mendell, J.T.; et al. Mir205hg is a long noncoding rna that regulates growth hormone and prolactin production in the anterior pituitary. Dev. Cell 2019, 49, 618–631. [Google Scholar] [CrossRef]
- Han, D.X.; Sun, X.L.; Xu, M.Q.; Chen, C.Z.; Jiang, H.; Gao, Y.; Yuan, B.; Zhang, J.B. Roles of differential expression of microrna-21-3p and microrna-433 in fsh regulation in rat anterior pituitary cells. Oncotarget 2017, 8, 36553–36565. [Google Scholar] [CrossRef] [PubMed]
- Han, D.X.; Sun, X.L.; Wang, C.J.; Yu, Z.W.; Zheng, Y.; Huang, Y.J.; Wang, W.H.; Jiang, H.; Gao, Y.; Yuan, B.; et al. Differentially expressed lncrna-m433s1 regulates fsh secretion by functioning as a mirna sponge in male rat anterior pituitary cells. Biol. Reprod. 2019, 101, 416–425. [Google Scholar] [CrossRef]
- Chen, S.W.; Zhu, J.; Ma, J.; Zhang, J.L.; Zuo, S.; Chen, G.W.; Wang, X.; Pan, Y.S.; Liu, Y.C.; Wang, P.Y. Overexpression of long non-coding rna h19 is associated with unfavorable prognosis in patients with colorectal cancer and increased proliferation and migration in colon cancer cells. Oncol. Lett. 2017, 14, 2446–2452. [Google Scholar] [CrossRef]
- Magdalena, M.; Yunzhe, Z.; Weijia, Z.; Kaixiang, C.; Chenyi, P.; Nathalie, L.; Mcdonald, J.F.; Bouhassira, E.E.; Yuhong, F. Histone h1.3 suppresses h19 noncoding rna expression and cell growth of ovarian cancer cells. Cancer Res. 2014, 74, 6463–6473. [Google Scholar]
- Lu, T.; Yu, C.; Ni, H.; Liang, W.; Yan, H.; Jin, W. Expression of the long non-coding rna h19 and malat-1 in growth hormone-secreting pituitary adenomas and its relationship to tumor behavior. Int. J. Dev. Neurosci. 2018, 67, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.R.; Yan, L.; Liu, Y.T.; Cao, L.; Guo, Y.H.; Zhang, Y.; Yao, H.; Cai, L.; Shang, H.B.; Rui, W.W.; et al. Inhibition of mtorc1 by lncrna h19 via disrupting 4e-bp1/raptor interaction in pituitary tumours. Nat. Commun. 2018, 9, 4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, G.; Gao, Y.; Zhao, C.; Li, X.; Zhang, F.; Jiang, C.; Wu, B. Lnc-snhg1 activates the tgfbr2/smad3 and rab11a/wnt/beta-catenin pathway by sponging mir-302/372/373/520 in invasive pituitary tumors. Cell Physiol. Biochem. 2018, 48, 1291–1303. [Google Scholar] [CrossRef]
- D’Angelo, D.; Mussnich, P.; Sepe, R.; Raia, M.; Del Vecchio, L.; Cappabianca, P.; Pellecchia, S.; Petrosino, S.; Saggio, S.; Solari, D.; et al. Rpsap52 lncrna is overexpressed in pituitary tumors and promotes cell proliferation by acting as mirna sponge for hmga proteins. J. Mol. Med. 2019, 97, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Zhang, Y.; Cui, H. Long noncoding rna ccat2 is activated by e2f1 and exerts oncogenic properties by interacting with pttg1 in pituitary adenomas. Am. J. Cancer Res. 2018, 8, 245–255. [Google Scholar] [PubMed]
- Li, Z.; Li, C.; Liu, C.; Yu, S.; Zhang, Y. Expression of the long non-coding rnas meg3, hotair, and malat-1 in non-functioning pituitary adenomas and their relationship to tumor behavior. Pituitary 2015, 18, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Chunharojrith, P.; Nakayama, Y.; Jiang, X.; Kery, R.E.; Ma, J.; De La Hoz Ulloa, C.S.; Zhang, X.; Zhou, Y.; Klibanski, A. Tumor suppression by meg3 lncrna in a human pituitary tumor derived cell line. Mol. Cell. Endocrinol. 2015, 416, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Li, C.; Xie, W.; Wang, Z.; Gao, H.; Cao, L.; Hao, L.; Zhang, Y. Long non-coding rna c5orf66-as1 is downregulated in pituitary null cell adenomas and is associated with their invasiveness. Oncol. Rep. 2017, 38, 1140–1148. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Tan, C.; Wen, Y.; Zhang, D.; Li, G.; Chang, L.; Su, J.; Wang, X. Foxp1-induced lncrna clrn1-as1 acts as a tumor suppressor in pituitary prolactinoma by repressing the autophagy via inactivating wnt/beta-catenin signaling pathway. Cell Death Dis. 2019, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, H.; Han, L.; Li, F.; Zhang, T.; Pang, J.; Feng, X.; Ren, C.; Mao, S.; Wang, F. Long noncoding rna expression profile changes associated with dietary energy in the sheep testis during sexual maturation. Sci. Rep. 2017, 7, 5180. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Li, F.; Wang, F.; Zhang, G.; Pang, J.; Ren, C.; Zhang, T.; Yang, H.; Wang, Z.; Zhang, Y. Genome-wide differential expression profiling of mrnas and lncrnas associated with prolificacy in hu sheep. Biosci. Rep. 2018, 38, BSR20171350. [Google Scholar] [CrossRef] [Green Version]
- La, Y.; Tang, J.; He, X.; Di, R.; Wang, X.; Liu, Q.; Zhang, L.; Zhang, X.; Zhang, J.; Hu, W.; et al. Identification and characterization of mrnas and lncrnas in the uterus of polytocous and monotocous small tail han sheep (ovis aries). PeerJ 2019, 7, e6938. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zhang, M.; Jin, Y.; Erdenee, S.; Hu, L.; Chen, H.; Cai, Y.; Lan, X. Comparative transcriptome profiling of mrna and lncrna related to tail adipose tissues of sheep. Front. Genet. 2018, 9, 365. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Wureli, H.; Ni, W.; Zhang, M.; Li, H.; Xu, Y.; Rizabek, K.; Bolatkhan, M.; Askar, D.; et al. Screening and evaluating of long non-coding rnas in prenatal and postnatal pituitary gland of sheep. Genomics 2019, 112, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, Z.; Yang, H.; Yao, X.; Yang, P.; Ren, C.; Wang, F.; Zhang, Y. Pituitary transcriptomic study reveals the differential regulation of lncrnas and mrnas related to prolificacy in different fecb genotyping sheep. Genes 2019, 10, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.X.; Sun, X.L.; Fu, Y.; Wang, C.J.; Liu, J.B.; Jiang, H.; Gao, Y.; Chen, C.Z.; Yuan, B.; Zhang, J.B. Identification of long non-coding rnas in the immature and mature rat anterior pituitary. Sci. Rep. 2017, 7, 17780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, F.; Li, F.; Ren, C.; Pang, J.; Wan, Y.; Wang, Z.; Feng, X.; Zhang, Y. Comprehensive analysis of long noncoding rna and mrna expression patterns in sheep testicular maturation. Biol. Reprod. 2018, 99, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Chan, C.K.K. Analysis of rna-seq data using tophat and cufflinks. In Plant Bioinformatics; Edwards, D., Ed.; Humana Press: New York, NY, USA, 2016; Volume 1374, pp. 339–361. [Google Scholar]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. Cpc: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Mao, T. Kegg as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.F.; Guo, X.H.; Du, M.; Cao, G.Q.; Yang, Q.C.; Pu, Z.D.; Wang, Z.Y.; Zhang, Q.; Li, M.; Jin, Y.S. Lncrna profiling of skeletal muscles in large white pigs and mashen pigs during development. J. Anim. Sci. 2017, 95, 4239. [Google Scholar] [CrossRef] [Green Version]
- Rotini, A.; Martínez-Sarrà, E.; Pozzo, E.; Sampaolesi, M. Interactions between micrornas and long non-coding rnas in cardiac development and repair. Pharmacol. Res. 2017, 127, S1043661817303572. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.E.; Lim, D.A. Forging our understanding of lncrnas in the brain. Cell Tissue Res. 2017, 371, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.R. Lncrna-map2k4 sequesters mir-199a to promote fgf1 expression and spinal cord neuron growth. Biochem. Biophys. Res. Commun. 2017, 490, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Li, H.; Du, B.; Cui, K.; Xing, Y.; Zhao, X.; Zai, S. Lncrna dancr promotes migration and invasion through suppression of lncrna-let in gastric cancer cells. Biosci. Rep. 2017, 37, BSR20171070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Ye, J.; Yang, C.; Zhang, K.; Li, X.; Luo, L.; Ding, J.; Li, Y.; Cao, H.; Ling, Y. Screening and evaluating of long noncoding rnas in the puberty of goats. BMC Genom. 2017, 18, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Ye, J.; Yang, C.; Luo, L.; Liu, Y.; Ding, J.; Zhang, Y.; Ling, Y.; Huang, W.; Zhang, X. Rna-seq analysis of lncrna-controlled developmental gene expression during puberty in goat & rat. BMC Genet. 2018, 19, 19. [Google Scholar]
- Ran, M.; Chen, B.; Li, Z.; Wu, M.; Liu, X.; He, C.; Zhang, S.; Li, Z. Systematic identification of long noncoding rnas in immature and mature porcine testes1. Biol. Reprod. 2016, 94, 77. [Google Scholar] [CrossRef]
- Ren, J.; Du, X.; Zeng, T.; Chen, L.; Shen, J.; Lu, L.; Hu, J. Divergently expressed gene identification and interaction prediction of long noncoding rna and mrna involved in duck reproduction. Anim. Reprod. Sci. 2017, 185, 8. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Ni, W.; Wang, D.; Hou, X.; Liu, Z.; Cao, Y.; Yao, Y.; Zhang, X.; Hu, S. Identification and comparison of micrornas in pituitary gland during prenatal and postnatal stages of sheep by deep sequencing. J. Genet. 2018, 97, 965–975. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Ma, Q.; Zhang, X.; Hu, S. Genome-wide analysis of circular rnas in prenatal and postnatal pituitary glands of sheep. Sci. Rep. 2017, 7, 16143. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Ramírez, L.C.; Trivellin, G.; Stratakis, C.A. Cyclic 3′,5′-adenosine monophosphate (camp) signaling in the anterior pituitary gland in health and disease. Mol. Cell. Endocrinol. 2018, 463, 72–86. [Google Scholar]
- Riccetti, L.; Sperduti, S.; Lazzaretti, C.; Casarini, L.; Simoni, M. The camp/pka pathway: Steroidogenesis of the antral follicular stage. Minerva Ginecol. 2018, 70, 516–624. [Google Scholar] [CrossRef] [PubMed]
- Lania, A.; Mantovani, G.; Spada, A. Camp pathway and pituitary tumorigenesis. Ann. Endocrinol. 2012, 73, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Lodge, E.J.; Russell, J.P.; Patist, A.L.; Francis-West, P.; Andoniadou, C.L. Expression analysis of the hippo cascade indicates a role in pituitary stem cell development. Front Physiol. 2016, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquale, C.D.; Gentilin, E.; Falletta, S.; Bellio, M.; Buratto, M.; degli Uberti, E.; Chiara Zatelli, M. Pi3k/akt/mtor pathway involvement in regulating growth hormone secretion in a rat pituitary adenoma cell line. Endocrine 2018, 60, 308–316. [Google Scholar] [CrossRef]
- Choi, Y.S.; Lee, H.J.; Ku, C.R.; Cho, Y.H.; Seo, M.R.; Lee, Y.J.; Lee, E.J. Foxo1 is a negative regulator of fshbeta gene expression in basal and gnrh-stimulated conditions in female. Endocrinology 2014, 155, 2277–2286. [Google Scholar] [CrossRef] [Green Version]
- St John, M.A.; Tao, W.; Fei, X.; Fukumoto, R.; Carcangiu, M.L.; Brownstein, D.G.; Parlow, A.F.; McGrath, J.; Xu, T. Mice deficient of lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 1999, 21, 182. [Google Scholar] [CrossRef]
- Ma, Y.; Chai, L.; Cortez, S.C.; Stopa, E.G.; Steinhoff, M.M.; Ford, D.; Morgan, J.; Maizel, A.L. Sall1 expression in the human pituitary-adrenal/gonadal axis. J. Endocrinol. 2002, 173, 437–448. [Google Scholar] [CrossRef]
- Herrero, M.J.; Lepesant, J.M. Daily and seasonal expression of clock genes in the pituitary of the european sea bass (dicentrarchus labrax). Gen. Comp. Endocrinol. 2014, 208, 30–38. [Google Scholar] [CrossRef]
- Moran, T.B.; Goldberg, L.B.; Serviss, S.L.; Raetzman, L.T. Numb deletion in pomc-expressing cells impairs pituitary intermediate lobe cell adhesion, progenitor cell localization, and neuro-intermediate lobe boundary formation. Mol. Endocrinol. 2011, 25, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Thakur, M.; Taha, D.; Misra, V.K. A case of congenital hypopituitarism associated with a 1p31 microdeletion: A possible role forleprandjak1. J. Endocr. Soc. 2017, 1, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto-Yamauchi, N.; Terasaka, T.; Iwasaki, Y.; Otsuka, F. Interaction of pituitary hormones and expression of clock genes modulated by bone morphogenetic protein-4 and melatonin. Biochem. Biophys. Res. Commun. 2015, 459, 172–177. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Z.; Xue, J.; Guo, X.; Liang, S.; Liu, A. Effect of hypoxia on ddr1 expression in pituitary adenomas. Med. Sci. Monit. 2015, 21, 2433–2438. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Liang, H. Mir-132, mir-15a and mir-16 synergistically inhibit pituitary tumor cell proliferation, invasion and migration by targeting sox5. Cancer Lett. 2015, 356, 568–578. [Google Scholar]
- Kemiläinen, H.; Adam, M.; Mäki-Jouppila, J.; Damdimopoulou, P.; Damdimopoulos, A.E.; Kere, J.; Hovatta, O.; Laajala, T.D.; Aittokallio, T.; Adamski, J.; et al. The hydroxysteroid (17β) dehydrogenase family gene hsd17b12 is involved in the prostaglandin synthesis pathway, the ovarian function, and regulation of fertility. Endocrinology 2016, 157, en20161252. [Google Scholar] [CrossRef] [Green Version]
- Haizhong, F.; Lopez, G.Y.; Chung Kwon, K.; Angel, A.; Duncan, C.G.; Ryo, N.; Motoo, N.; Su, A.J.A.; Auron, P.E.; Hedberg, M.L. Egfr phosphorylation of dcbld2 recruits traf6 and stimulates akt-promoted tumorigenesis. J. Clin. Investig. 2014, 124, 3741–3756. [Google Scholar]
- Wang, H.; Zhang, L.; Cao, J.; Wu, M.; Ma, X.; Liu, Z.; Liu, R.; Zhao, F.; Wei, C.; Du, L. Genome-wide specific selection in three domestic sheep breeds. PLoS ONE 2015, 10, e0128688. [Google Scholar] [CrossRef] [Green Version]
- Dettori, M.L.; Pazzola, M.; Paschino, P.; Amills, M.; Vacca, G.M. Association between the ghr, ghrhr, and igf1 gene polymorphisms and milk yield and quality traits in sarda sheep. J. Dairy Sci. 2018, 101, 9978–9986. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Han, B.; Zhu, W.; Cheng, T.; Fan, M.; Wu, J.; Yang, Y.; Zhu, H.; Si, J.; Lyu, Q. Polymorphism in the alternative donor site of the cryptic exon of lhcgr: Functional consequences and associations with testosterone level. Sci. Rep. 2017, 7, 45699. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNA | UP/DOWN | Targeted Gene | Relationship | Mainly Pathways Enrichment |
|---|---|---|---|---|
| TCONS_00066406 | UP | HSD17B12 | antisense | Steroid hormone biosynthesis |
| XR_003591760.1 | DOWN | DCBLD2 | antisense | regulation of growth; regulation of cell growth |
| TCONS_00084471 | DOWN | PDPK1 | cis, upstream | PI3K-Akt signaling pathway; mTOR signaling pathway; AMPK signaling pathway; Apoptosis; FoxO signaling pathway; Thyroid hormone signaling pathway; PPAR signaling pathway |
| TCONS_00032215 | DOWN | GPX3 | cis, downstream | Thyroid hormone synthesis; cellular process |
| TCONS_00045021 | UP | DLL1 | cis, upstream | Notch signaling pathway |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Ma, J.; Wang, Z.; Yao, X.; Zhao, J.; Zhao, X.; Wang, F.; Zhang, Y. Genome-Wide Analysis and Function Prediction of Long Noncoding RNAs in Sheep Pituitary Gland Associated with Sexual Maturation. Genes 2020, 11, 320. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030320
Yang H, Ma J, Wang Z, Yao X, Zhao J, Zhao X, Wang F, Zhang Y. Genome-Wide Analysis and Function Prediction of Long Noncoding RNAs in Sheep Pituitary Gland Associated with Sexual Maturation. Genes. 2020; 11(3):320. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030320
Chicago/Turabian StyleYang, Hua, Jianyu Ma, Zhibo Wang, Xiaolei Yao, Jie Zhao, Xinyue Zhao, Feng Wang, and Yanli Zhang. 2020. "Genome-Wide Analysis and Function Prediction of Long Noncoding RNAs in Sheep Pituitary Gland Associated with Sexual Maturation" Genes 11, no. 3: 320. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030320