A Novel Germline MLH1 In-Frame Deletion in a Slovenian Lynch Syndrome Family Associated with Uncommon Isolated PMS2 Loss in Tumor Tissue

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
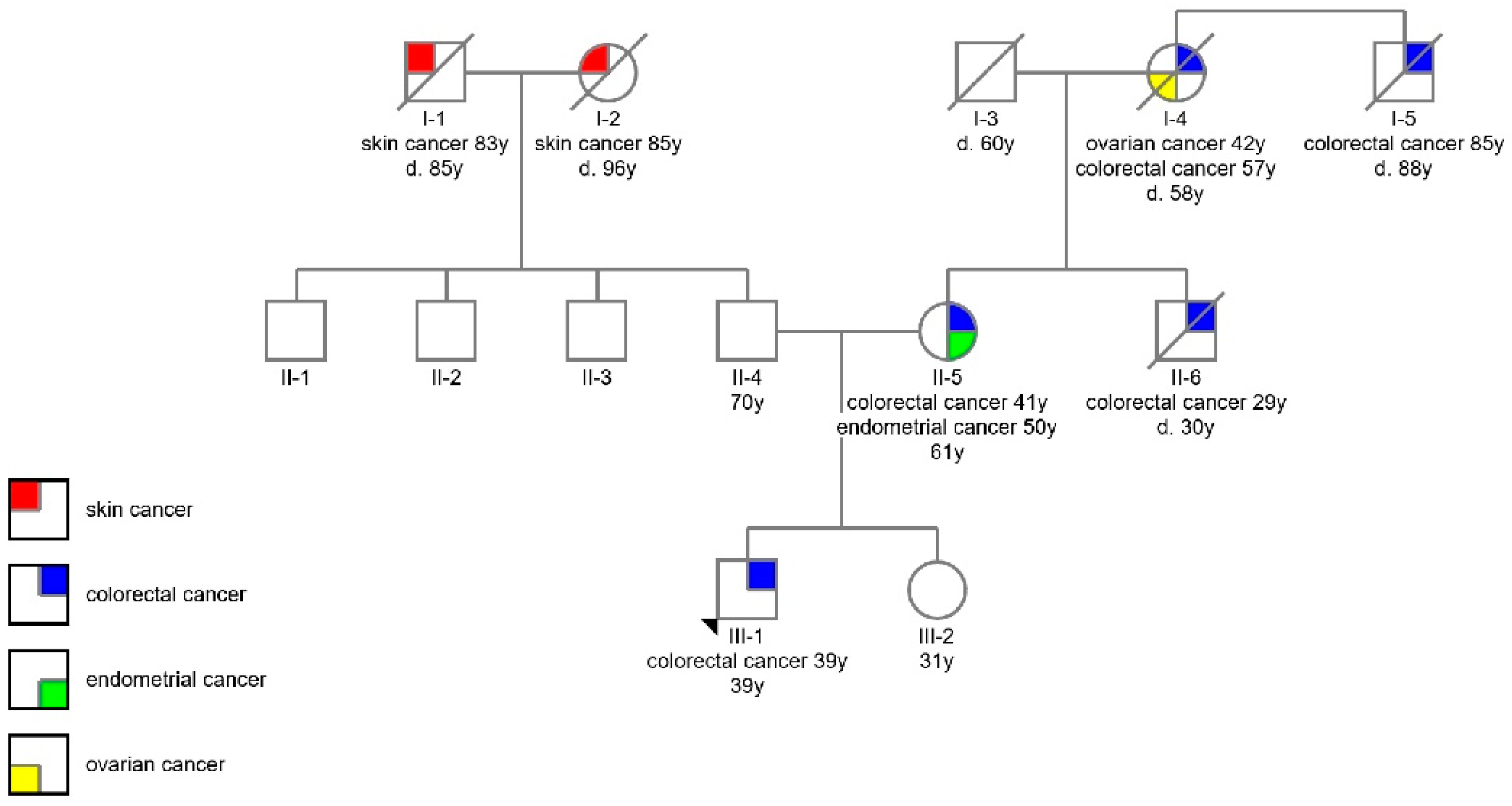
2.1. Participants
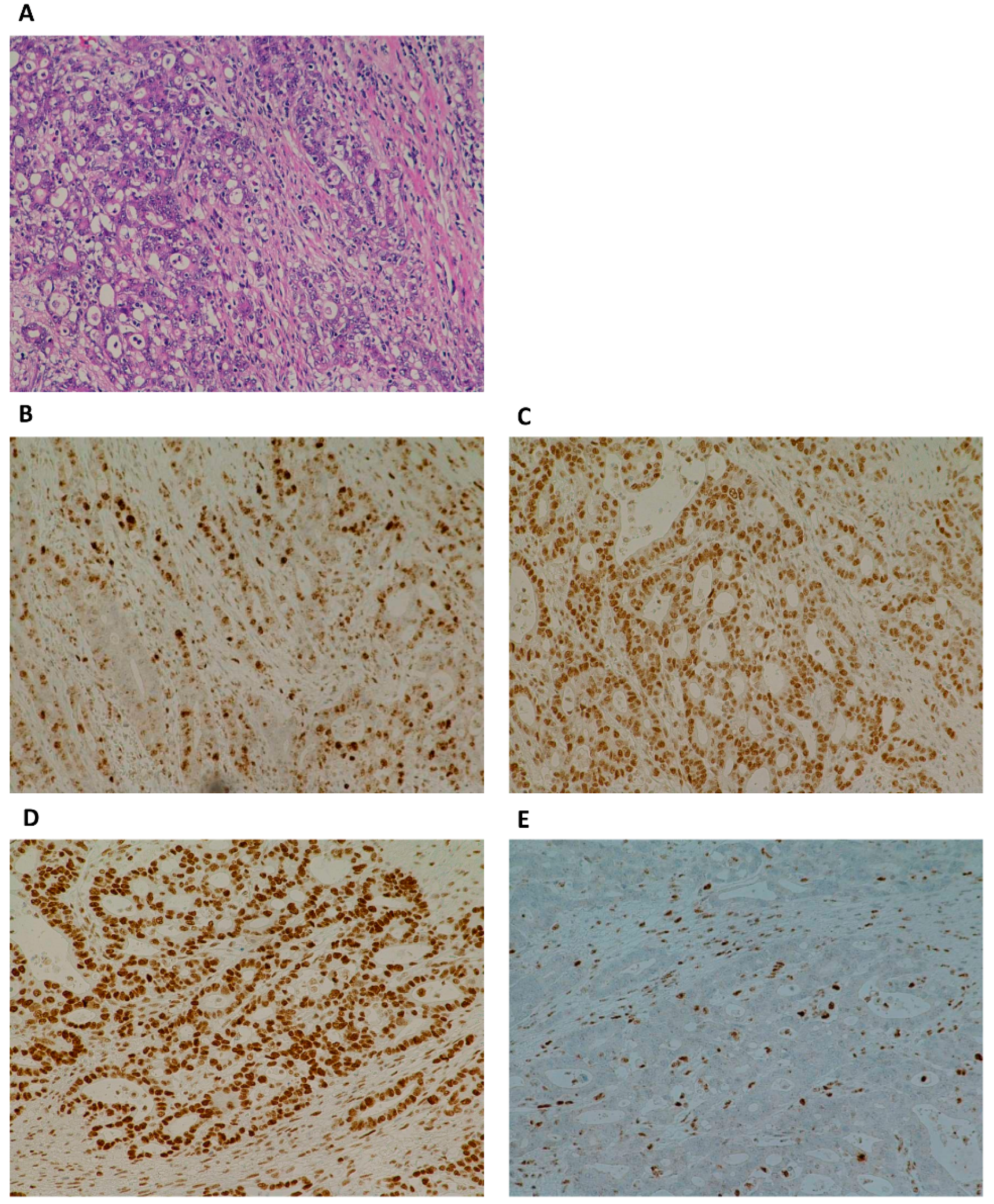
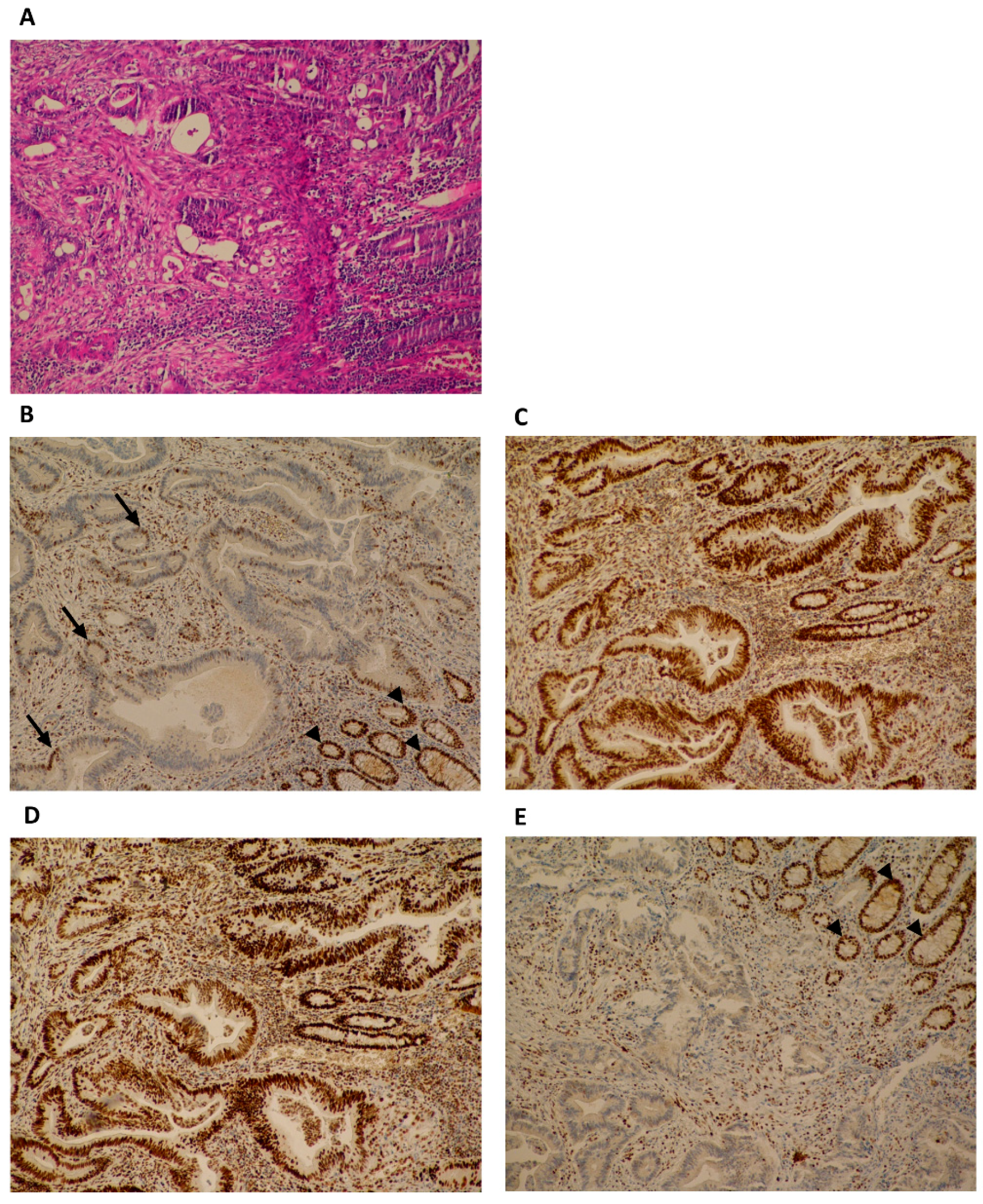
2.2. Immunohistochemistry
2.3. DNA Isolation
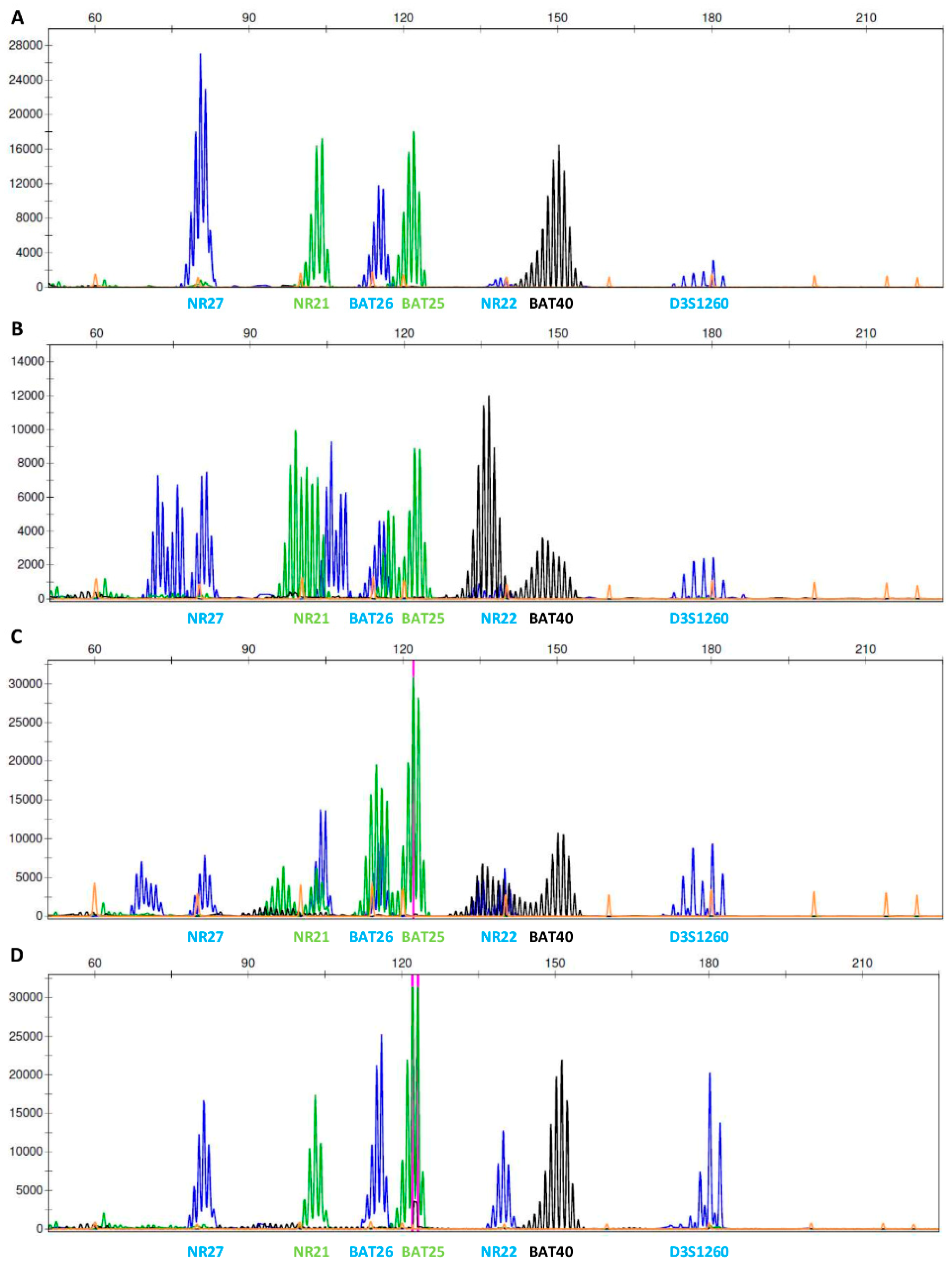
2.4. Microsatellite Instability
2.5. Next Generation Sequencing
2.5.1. Genotyping for Germline Alterations
2.5.2. Genotyping for Sporadic Alterations
2.6. MLPA
2.7. Sanger Sequencing
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Lynch, P.M. Hyperplastic polyposis: Semantics, biology, and endoscopy. Gut 2010, 59, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895-2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Young, J.P.; Lindor, N.M.; Tucker, K.M.; Ahnen, D.J.; Young, G.P.; Buchanan, D.D.; Clendenning, M.; Giles, G.G.; Winship, I.; et al. Colorectal and Other Cancer Risks for Carriers and Noncarriers From Families With a DNA Mismatch Repair Gene Mutation: A Prospective Cohort Study. J. Clin. Oncol. 2012, 30, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; de la Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2 -Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef] [Green Version]
- Kastrinos, F.; Stoffel, E.M. History, Genetics, and Strategies for Cancer Prevention in Lynch Syndrome. Clin. Gastroenterol. Hepatol. 2014, 12, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Markowitz, A.J.; Chung, D.C.; Mayer, R.J.; Regenbogen, S.E.; Blanco, A.M.; Bray, T.; Cooper, G.; et al. NCCN Guidelines Insights: Colorectal Cancer Screening, Version 1.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 939–949. [Google Scholar] [CrossRef]
- Gupta, S.; Provenzale, D.; Llor, X.; Halverson, A.L.; Grady, W.; Chung, D.C.; Haraldsdottir, S.; Markowitz, A.J.; Slavin, T.P.; Hampel, H.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J. Natl. Compr. Canc. Netw. 2019, 17, 1032–1041. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.H.; Daniels, M. Endometrial and ovarian cancer in women with Lynch syndrome: Update in screening and prevention. Fam. Cancer 2013, 12, 273–277. [Google Scholar] [CrossRef]
- Schmeler, K.M.; Lynch, H.T.; Chen, L.; Munsell, M.F.; Soliman, P.T.; Clark, M.B.; Daniels, M.S.; White, K.G.; Boyd-Rogers, S.G.; Conrad, P.G.; et al. Prophylactic Surgery to Reduce the Risk of Gynecologic Cancers in the Lynch Syndrome. N. Engl. J. Med. 2006, 354, 261–269. [Google Scholar] [CrossRef] [Green Version]
- van Lier, M.G.F.; Wagner, A.; van Leerdam, M.E.; Biermann, K.; Kuipers, E.J.; Steyerberg, E.W.; Dubbink, H.J.; Dinjens, W.N.M. A review on the molecular diagnostics of Lynch syndrome: A central role for the pathology laboratory. J. Cell. Mol. Med. 2010, 14, 181–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, A.E.; Hendriks, Y.M.C.; Kleibeuker, J.H.; de Boer, S.Y.; Cats, A.; Griffioen, G.; Nagengast, F.M.; Nelis, F.G.; Rookus, M.A.; Vasen, H.F.A. Decrease in Mortality in Lynch Syndrome Families Because of Surveillance. Gastroenterology 2006, 130, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Stupart, D.A.; Goldberg, P.A.; Algar, U.; Ramesar, R. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Color. Dis. 2009, 11, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Moreno-ortiz, J.M.; Ayala-madrigal, M.D.L.; Corona-rivera, J.R.; Centeno-flores, M.; Maciel-gutiérrez, V.; Franco-topete, R.A.; Armendáriz-borunda, J.; Hotchkiss, E.; Pérez-carbonell, L.; Rhees, J.; et al. Novel Mutations in MLH1 and MSH2 Genes in Mexican Patients with Lynch Syndrome. Gastroenterol. Res. Pract. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sijmons, R.H.; Hofstra, R.M.W. Review: Clinical aspects of hereditary DNA Mismatch repair gene mutations. DNA Repair (Amst). 2016, 38, 155–162. [Google Scholar] [CrossRef]
- Lagerstedt Robinson, K.; Liu, T.; Vandrovcova, J.; Halvarsson, B.; Clendenning, M.; Frebourg, T.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Peltomäki, P.; et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J. Natl. Cancer Inst. 2007, 99, 291–299. [Google Scholar] [CrossRef]
- Martín-López, J.V.; Fishel, R. The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Fam. Cancer 2013, 12, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges. Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [Green Version]
- Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Cox, V.L.; Saeed Bamashmos, A.A.; Foo, W.C.; Gupta, S.; Yedururi, S.; Garg, N.; Kang, H.C. Lynch Syndrome: Genomics Update and Imaging Review. RadioGraphics 2018, 38, 483–499. [Google Scholar] [CrossRef]
- Sjursen, W.; Mcphillips, M.; Scott, R.J.; Talseth-palmer, B.A. Lynch syndrome mutation spectrum in New South Wales, Australia, including 55 novel mutations. Molecular Gene. Genomic Med. 2016, 1, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyström-Lahti, M.; Wu, Y.; Moisio, A.L.; Hofstra, R.M.; Osinga, J.; Mecklin, J.P.; Järvinen, H.J.; Leisti, J.; Buys, C.H.; de la Chapelle, A.; et al. DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum. Mol. Genet. 1996, 5, 763–769. [Google Scholar] [CrossRef]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Marqués-Lespier, J.M.; Diaz-Algorri, Y.; Gonzalez-Pons, M.; Cruz-Correa, M. Report of a Novel Mutation in MLH1 Gene in a Hispanic Family from Puerto Rico Fulfilling Classic Amsterdam Criteria for Lynch Syndrome. Gastroenterol. Res. Pract. 2014, 2014, 527946. [Google Scholar] [CrossRef] [Green Version]
- Lindor, N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-Vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.A.; et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J. Clin. Oncol. 2002, 20, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Raevaara, T.E. Description and functional analysis of a novel in frame mutation linked to hereditary non-polyposis colorectal cancer. J. Med. Genet. 2002, 39, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Dudley, B.; Brand, R.E.; Thull, D.; Bahary, N.; Nikiforova, M.N.; Pai, R.K. Germline MLH1 Mutations Are Frequently Identified in Lynch Syndrome Patients With Colorectal and Endometrial Carcinoma Demonstrating Isolated Loss of PMS2 Immunohistochemical Expression. Am. J. Surg. Pathol. 2015, 39, 1114–1120. [Google Scholar] [CrossRef]
- Alpert, L.; Pai, R.K.; Srivastava, A.; McKinnon, W.; Wilcox, R.; Yantiss, R.K.; Arcega, R.; Wang, H.L.; Robert, M.E.; Liu, X.; et al. Colorectal Carcinomas With Isolated Loss of PMS2 Staining by Immunohistochemistry. Arch. Pathol. Lab. Med. 2018, 142, 523–528. [Google Scholar] [CrossRef]
- Pagin, A.; Zerimech, F.; Leclerc, J.; Wacrenier, A.; Lejeune, S.; Descarpentries, C.; Escande, F.; Porchet, N. Evaluation of a new panel of six mononucleotide repeat markers for the detection of DNA mismatch repair-deficient tumours. Br. J. Cancer 2013, 2079–2087. [Google Scholar] [CrossRef]
- Setrajcic Dragos, V.; Blatnik, A.; Klancar, G.; Stegel, V.; Krajc, M.; Blatnik, O.; Novakovic, S. Two Novel NF1 Pathogenic Variants Causing the Creation of a New Splice Site in Patients With Neurofibromatosis Type I. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Dai, J.; Wang, D.; Lu, H.; Dai, H.; Ye, H.; Gu, J.; Chen, S.; Huang, B. Assessment of tumor mutation burden calculation from gene panel sequencing data. Onco. Targets. Ther. 2019, Volume 12, 3401–3409. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D’Haene, N. Methods of measurement for tumor mutational burden in tumor tissue. Transl. Lung Cancer Res. 2018, 7, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in Eukaryotic Mismatch Repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd, A.B.; Palama, B.; Nelson, S.E.; Tomer, G.; Nguyen, M.; Huo, X.; Buermeyer, A.B. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst). 2006, 5, 347–361. [Google Scholar] [CrossRef]
- D’Arcy, B.M.; Blount, J.; Prakash, A. Biochemical and structural characterization of two variants of uncertain significance in the PMS2 gene. Hum. Mutat. 2019, 40, 458–471. [Google Scholar] [CrossRef]
- Richman, S. Deficient mismatch repair: Read all about it (Review). Int. J. Oncol. 2015, 47, 1189–1202. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.; Lindor, N.M.; Burgart, L.J.; Smalley, R.; Leontovich, O.; French, A.; Goldberg, R.M.; Sargent, D.J.; Jass, J.R.; Hopper, J.L.; et al. Isolated loss of PMS2 expression in colorectal cancers: Frequency, patient age, and familial aggregation. Clin. Cancer Res. 2005, 11, 6466–6471. [Google Scholar] [CrossRef] [Green Version]
- Guerrette, S.; Acharya, S.; Fishel, R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J. Biol. Chem. 1999, 274, 6336–6341. [Google Scholar] [CrossRef] [Green Version]
- Hinrichsen, I.; Weßbecher, I.M.; Huhn, M.; Passmann, S.; Zeuzem, S.; Plotz, G.; Biondi, R.M.; Brieger, A. Phosphorylation-dependent signaling controls degradation of DNA mismatch repair protein PMS2. Mol. Carcinog. 2017, 56, 2663–2668. [Google Scholar] [CrossRef]
- de Jong, A.E.; van Puijenbroek, M.; Hendriks, Y.; Tops, C.; Wijnen, J.; Ausems, M.G.E.M.; Meijers-Heijboer, H.; Wagner, A.; van Os, T.A.M.; Bröcker-Vriends, A.H.J.T.; et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin. Cancer Res. 2004, 10, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Rosty, C.; Clendenning, M.; Walsh, M.D.; Eriksen, S.V.; Southey, M.C.; Winship, I.M.; Macrae, F.A.; Boussioutas, A.; Poplawski, N.K.; Parry, S.; et al. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open 2016, 6, e010293. [Google Scholar] [CrossRef] [Green Version]
- Borelli, I.; Casalis Cavalchini, G.C.; Del Peschio, S.; Micheletti, M.; Venesio, T.; Sarotto, I.; Allavena, A.; Delsedime, L.; Barberis, M.A.; Mandrile, G.; et al. A founder MLH1 mutation in Lynch syndrome families from Piedmont, Italy, is associated with an increased risk of pancreatic tumours and diverse immunohistochemical patterns. Fam. Cancer 2014, 13, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.C.C.; Torrezan, G.T.; Ferreira, J.R.O.; Oliveira, L.P.; Begnami, M.D.F.S.; Aguiar, S.; Carraro, D.M. Germline Mutations in MLH1 Leading to Isolated Loss of PMS2 Expression in Lynch Syndrome: Implications for Diagnostics in the Clinic. Am. J. Surg. Pathol. 2017, 41, 861–864. [Google Scholar] [CrossRef]
- Zighelboim, I.; Powell, M.A.; Babb, S.A.; Whelan, A.J.; Schmidt, A.P.; Clendenning, M.; Senter, L.; Thibodeau, S.N.; de la Chapelle, A.; Goodfellow, P.J. Epitope-positive truncating MLH1 mutation and loss of PMS2: Implications for IHC-directed genetic testing for Lynch syndrome. Fam. Cancer 2009, 8, 501–504. [Google Scholar] [CrossRef] [Green Version]
- Halvarsson, B.; Lindblom, A.; Rambech, E.; Lagerstedt, K.; Nilbert, M. The added value of PMS2 immunostaining in the diagnosis of hereditary nonpolyposis colorectal cancer. Fam. Cancer 2006, 5, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Hechtman, J.F.; Rana, S.; Middha, S.; Stadler, Z.K.; Latham, A.; Benayed, R.; Soslow, R.; Ladanyi, M.; Yaeger, R.; Zehir, A.; et al. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated with missense mutations in mismatch repair genes. Mod. Pathol. 2019. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Fardaei, M.; Lankarani, K.B.; Miryounesi, M. Segregation of a novel MLH1 mutation in an Iranian Lynch syndrome family. Gene 2015, 570, 304–305. [Google Scholar] [CrossRef]
- Tomsic, J.; Liyanarachchi, S.; Hampel, H.; Morak, M.; Thomas, B.C.; Raymond, V.M.; Chittenden, A.; Schackert, H.K.; Gruber, S.B.; Syngal, S.; et al. An American founder mutation in MLH1. Int. J. Cancer 2012, 130, 2088–2095. [Google Scholar] [CrossRef] [Green Version]
- Erffelinck, M.-L.; Ribeiro, B.; Perassolo, M.; Pauwels, L.; Pollier, J.; Storme, V.; Goossens, A. A user-friendly platform for yeast two-hybrid library screening using next generation sequencing. PLoS ONE 2018, 13, e0201270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.J.; Rasmussen, L.J.; Hofstra, R.; Monteiro, A.N.A.; Greenblatt, M.S.; de Wind, N.; IARC Unclassified Genetic Variants Working Group. Assessment of functional effects of unclassified genetic variants. Hum. Mutat. 2008, 29, 1314–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Member | Tumor as a Result of MLH1 Variant Causing Lynch Syndrome | Material | Tumor Cells (%) | IHC (Expression) | MSI Status | MLH1 Variant Allele Fraction (%) | MLPA (PMS2 Gene) | TMB (mut/Mb) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor Sample | Non-Tumor Sample | MLH1 (%) | MSH2 (%) | MSH6 (%) | PMS2 (%) | |||||||
| Proband III-1 | yes | / | blood | 0 | N/A | N/A | N/A | N/A | MSI-S | 50 | wt | N/A |
| cecum | / | 85–90 | 100 | 100 | 100 | 0 | MSI-H | 55 | N/A | 22 | ||
| III-2 | no | / | blood | 0 | N/A | N/A | N/A | N/A | N/A | wt | N/A | N/A |
| II-5 | yes | / | blood | 0 | N/A | N/A | N/A | N/A | MSI-S | 50 | wt | N/A |
| / | lymph node nearby colon | 0 | N/A | N/A | N/A | N/A | MSI-S | 20 | N/A | N/A | ||
| / | ovary | 0 | N/A | N/A | N/A | N/A | MSI-S | 50 | N/A | N/A | ||
| ileum | / | 75 | 5 | 100 | 100 | 0 | MSI-H | 20 | N/A | N/A | ||
| endometrium (block 1) | / | 65 | ?1 | 100 | 100 | 0 | MSI-H | 50 | N/A | N/A | ||
| endometrium (block 2) | / | 75 | ?1 | 100 | 100 | 0 | MSI-H | 50 | N/A | N/A | ||
| II-6 | yes | / | cecum | 0 | N/A | N/A | N/A | N/A | MSI-S | 50 | N/A | N/A |
| cecum | / | 75 | 100 | 100 | 100 | 0 | MSI-H | 50 | N/A | N/A | ||
| I-5 | no | / | rectum | 0 | N/A | N/A | N/A | N/A | MSI-S | wt | N/A | N/A |
| rectum | / | 70 | 100 | 100 | 100 | 100 | MSI-S | wt | N/A | N/A | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klančar, G.; Blatnik, A.; Šetrajčič Dragoš, V.; Vogrič, V.; Stegel, V.; Blatnik, O.; Drev, P.; Gazič, B.; Krajc, M.; Novaković, S. A Novel Germline MLH1 In-Frame Deletion in a Slovenian Lynch Syndrome Family Associated with Uncommon Isolated PMS2 Loss in Tumor Tissue. Genes 2020, 11, 325. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030325
Klančar G, Blatnik A, Šetrajčič Dragoš V, Vogrič V, Stegel V, Blatnik O, Drev P, Gazič B, Krajc M, Novaković S. A Novel Germline MLH1 In-Frame Deletion in a Slovenian Lynch Syndrome Family Associated with Uncommon Isolated PMS2 Loss in Tumor Tissue. Genes. 2020; 11(3):325. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030325
Chicago/Turabian StyleKlančar, Gašper, Ana Blatnik, Vita Šetrajčič Dragoš, Vesna Vogrič, Vida Stegel, Olga Blatnik, Primož Drev, Barbara Gazič, Mateja Krajc, and Srdjan Novaković. 2020. "A Novel Germline MLH1 In-Frame Deletion in a Slovenian Lynch Syndrome Family Associated with Uncommon Isolated PMS2 Loss in Tumor Tissue" Genes 11, no. 3: 325. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030325