A Comparison between Hi-C and 10X Genomics Linked Read Sequencing for Whole Genome Phasing in Hanwoo Cattle

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Sample
2.2. 10X Genomics Sequencing and Analysis
2.3. Hi-C Chromosome Conformation Captured Reads Sequencing
2.4. Read Assembly and Haplotype Phasing
2.5. Metrics for Comparing Phasing Performance between the Platforms
3. Results and Discussion
3.1. Sequencing and Variant Calling
3.2. Genome-Scale Haplotype Phasing
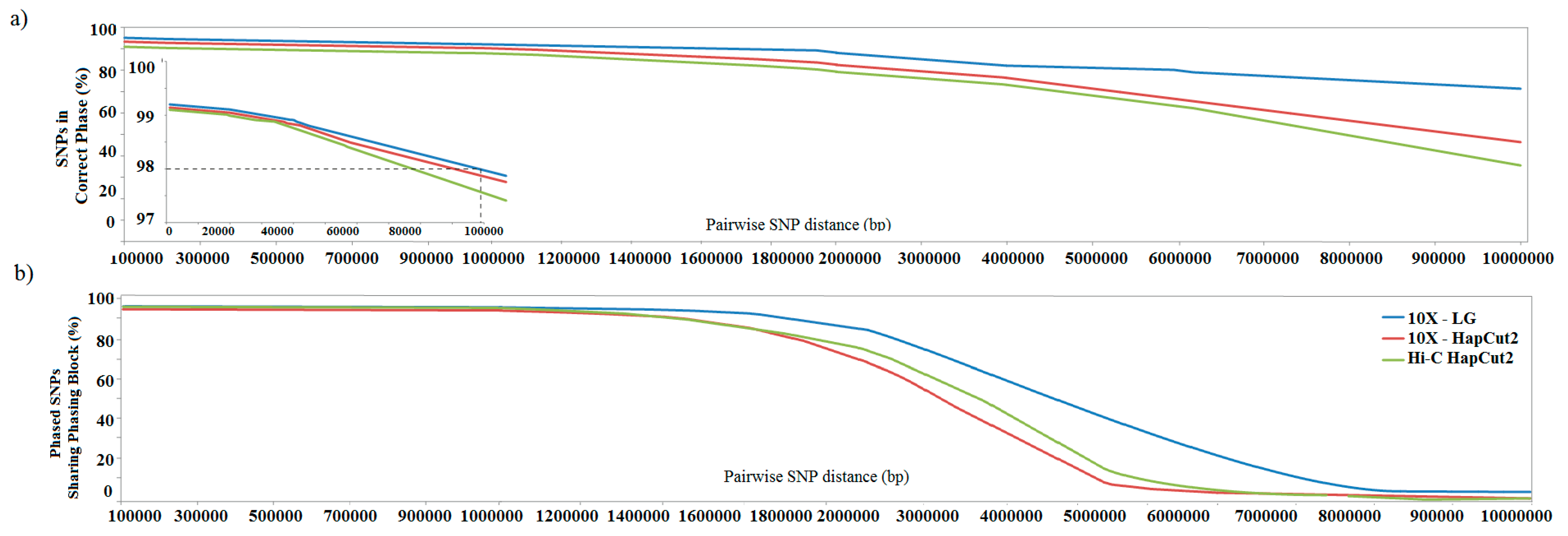
3.3. Estimating Accuracy of Haplotype Phasing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef]
- Hayes, B.J.; Daetwyler, H.D. 1000 Bull Genomes project to map simple and complex genetic traits in cattle: Applications and outcomes. Ann. Rev. Anim. Biosci. 2019, 7, 89–102. [Google Scholar] [CrossRef]
- Snyder, M.W.; Adey, A.; Kitzman, J.O.; Shendure, J. Haplotype-resolved genome sequencing: Experimental methods and applications. Nat. Rev. Genet. 2015, 16, 344. [Google Scholar] [CrossRef]
- Ben-Elazar, S.; Chor, B.; Yakhini, Z. Extending partial haplotypes to full genome haplotypes using chromosome conformation capture data. Bioinformatics 2016, 32, i559–i566. [Google Scholar] [CrossRef]
- Ramaker, R.C.; Savic, D.; Hardigan, A.A.; Newberry, K.; Cooper, G.M.; Myers, R.M.; Cooper, S.J. A genome-wide interactome of DNA-associated proteins in the human liver. Genome Res. 2017, 27, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Huddleston, J.; Eichler, E.E. An incomplete understanding of human genetic variation. Genetics 2016, 202, 1251–1254. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P.; Kirkness, E.; Telenti, A.; Schork, N.J. Comparison of phasing strategies for whole human genomes. PLoS Genet. 2018, 14, e1007308. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, J.A.; Kim, J.; Hamilton, J.P.; Wu, M.; Irber, L.C.; Maddamsetti, R.; Briney, B.; Newton, L.; Burton, D.R.; Brown, C.T. Haplotype-phased synthetic long reads from short-read sequencing. PLoS ONE 2016, 11, e0147229. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X. The potential and challenges of nanopore sequencing. In Nanoscience and Technology: A Collection of Reviews from Nature Journals; World Scientific: Singapore, 2010; pp. 261–268. [Google Scholar]
- Berlin, K.; Koren, S.; Chin, C.-S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, R.C.; Taylor, R.W.; Blauwkamp, T.A.; Kelley, J.L.; Kertesz, M.; Pushkarev, D.; Petrov, D.A.; Fiston-Lavier, A.-S. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE 2014, 9, e106689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Dixon, J.R.; Bansal, V.; Ren, B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat. Biotechnol. 2013, 31, 1111. [Google Scholar] [CrossRef] [PubMed]
- Bansal, V.; Bafna, V. HapCUT: An efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics 2008, 24, i153–i159. [Google Scholar] [CrossRef] [Green Version]
- Edge, P.; Bafna, V.; Bansal, V. HapCUT2: Robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 2017, 27, 801–812. [Google Scholar] [CrossRef]
- Shin, G.; Greer, S.U.; Xia, L.C.; Lee, H.; Zhou, J.; Boles, T.C.; Ji, H.P. Assembly of Mb-size genome segments from linked read sequencing of CRISPR DNA targets. bioRxiv 2018. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Zhao, Q.; Ming, R.; Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 2019, 5, 833–845. [Google Scholar] [CrossRef]
- Delaneau, O.; Marchini, J.; Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 2012, 9, 179–181. [Google Scholar] [CrossRef]
- Genomics, X. Sample Preparation Demonstated Protocol. 2020. Available online: https://assets.ctfassets.net/an68im79xiti/6PoCPM1BUQmkcw4SK8AGi2/ae196e362b118842eea7cd73a46c02f7/CG00019_SamplePrepDemonstratedProtocol_-_DNAQC_RevB.pdf (accessed on 21 December 2019).
- Genomics, X. Genome Reagents Kits v2 User Guide. 2020. Available online: https://assets.ctfassets.net/an68im79xiti/1Jw6vQfW1GOGuO0AsS2gM8/61866afe8c8af5e0eecf6a3d890f58aa/CG00043_GenomeReagentKitv2UserGuide_RevB.pdf (accessed on 21 December 2019).
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, G.; Dan, C.; Xiao, S.; Guo, W.; Huang, P.; Xiong, Y.; Wu, J.; He, Y.; Zhang, J.; Li, X. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. GigaScience 2018, 7, giy120. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, V.; Halpern, A.L.; Axelrod, N.; Bafna, V. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 2008, 18, 1336–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Kuleshov, V.; Xie, D.; Chen, R.; Pushkarev, D.; Ma, Z.; Blauwkamp, T.; Kertesz, M.; Snyder, M. Whole-genome haplotyping using long reads and statistical methods. Nat. Biotechnol. 2014, 32, 261. [Google Scholar] [CrossRef] [Green Version]
- Duitama, J.; McEwen, G.K.; Huebsch, T.; Palczewski, S.; Schulz, S.; Verstrepen, K.; Suk, E.-K.; Hoehe, M.R. Fosmid-based whole genome haplotyping of a HapMap trio child: Evaluation of Single Individual Haplotyping techniques. Nucleic Acids Res. 2011, 40, 2041–2053. [Google Scholar] [CrossRef] [Green Version]
- Amini, S.; Pushkarev, D.; Christiansen, L.; Kostem, E.; Royce, T.; Turk, C.; Pignatelli, N.; Adey, A.; Kitzman, J.O.; Vijayan, K. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet. 2014, 46, 1343. [Google Scholar] [CrossRef]
- Miar, Y.; Sargolzaei, M.; Schenkel, F.S. A comparison of different algorithms for phasing haplotypes using Holstein cattle genotypes and pedigree data. J. Dairy Sci. 2017, 100, 2837–2849. [Google Scholar] [CrossRef] [Green Version]
- Braz, C.U.; Taylor, J.F.; Bresolin, T.; Espigolan, R.; Feitosa, F.L.; Carvalheiro, R.; Baldi, F.; Lucia, G.; De Oliveira, H.N. Sliding window haplotype approaches overcome single SNP analysis limitations in identifying genes for meat tenderness in Nelore cattle. BMC Genet. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Zagury, J.-F.; Robinson, M.R.; Marchini, J.L.; Dermitzakis, E.T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, R.; Davies, R.W.; Heger, A.; Pagnamenta, A.T.; de Cesare, M.; Oikkonen, L.E.; Parkes, D.; Freeman, C.; Dhalla, F.; Patel, S.Y. Sequencing of human genomes with nanopore technology. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| 10X Genomics | Hi-C | |
|---|---|---|
| Total Reads | 790,643,590 | 441,889,616 |
| Mapped Reads | 756,645,916 (95.7%) | 438,995,090 (99.2%) |
| Q30 (%) | 92.50% | 93.00% |
| Mean Depth | 37.9X | 21.2X |
| 10X Genomics | Hi-C | |||
|---|---|---|---|---|
| SNPs | Indels | SNPs | Indels | |
| Total | 8,670,477 | 1,749,472 | 7,132,127 | 1,753,086 |
| Homozygous (%) | 2,590,042 (30%) | 507,761 (29%) | 2,170,128 (30%) | 417,903 (24%) |
| Heterozygous (%) | 6,080,435 (70%) | 1,241,711 (71%) | 4,397,837 (70%) | 1,335,183 (76%) |
| Sequencing Platform—Phasing Method | Metrics for Phasing Performance | |
|---|---|---|
| 10X-LG | Total SNPs Phased | 7,766,580 |
| % of SNPs Phased | 89.57 | |
| QAN50 (bp) | 1,249,365 | |
| SER * (%) | 0.07 | |
| 10X–HapCut2 | Total SNPs Phased | 5,836,541 |
| % of SNPs Phased | 67.31 | |
| QAN50 (bp) | 541,912 | |
| SER * (%) | 0.16 | |
| Hi-C–HapCut2 | Total SNPs Phased | 3,687,511 |
| % of SNPs Phased | 51.65 | |
| QAN50 (bp) | 1,034,586 | |
| SER *(%) | 0.24 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srikanth, K.; Park, J.-E.; Lim, D.; Cha, J.; Cho, S.-R.; Cho, I.-C.; Park, W. A Comparison between Hi-C and 10X Genomics Linked Read Sequencing for Whole Genome Phasing in Hanwoo Cattle. Genes 2020, 11, 332. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030332
Srikanth K, Park J-E, Lim D, Cha J, Cho S-R, Cho I-C, Park W. A Comparison between Hi-C and 10X Genomics Linked Read Sequencing for Whole Genome Phasing in Hanwoo Cattle. Genes. 2020; 11(3):332. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030332
Chicago/Turabian StyleSrikanth, Krishnamoorthy, Jong-Eun Park, Dajeong Lim, Jihye Cha, Sang-Rae Cho, In-Cheol Cho, and Woncheoul Park. 2020. "A Comparison between Hi-C and 10X Genomics Linked Read Sequencing for Whole Genome Phasing in Hanwoo Cattle" Genes 11, no. 3: 332. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030332