Quantitative Trait Loci (QTLs) Associated with Microspore Culture in Raphanus sativus L. (Radish)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genomic DNA Extraction
2.2. Microspore Culture and Regeneration
2.3. Genotyping-by-Sequencing (GBS) Library and Illumina Sequencing
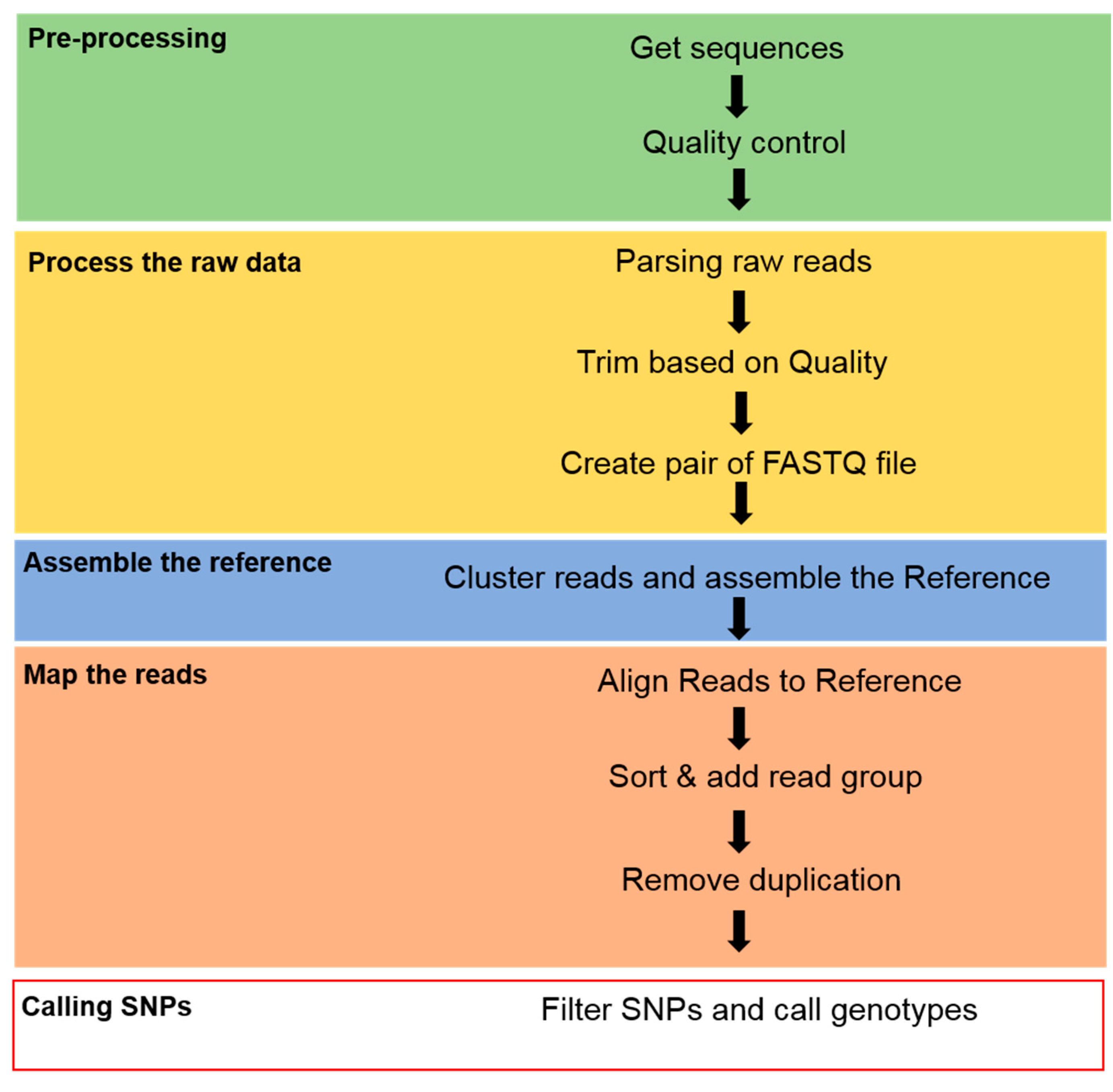
2.4. Sequence Data Analysis and SNP Calling
2.5. Linkage Map Construction
2.6. QTL Analysis
3. Results
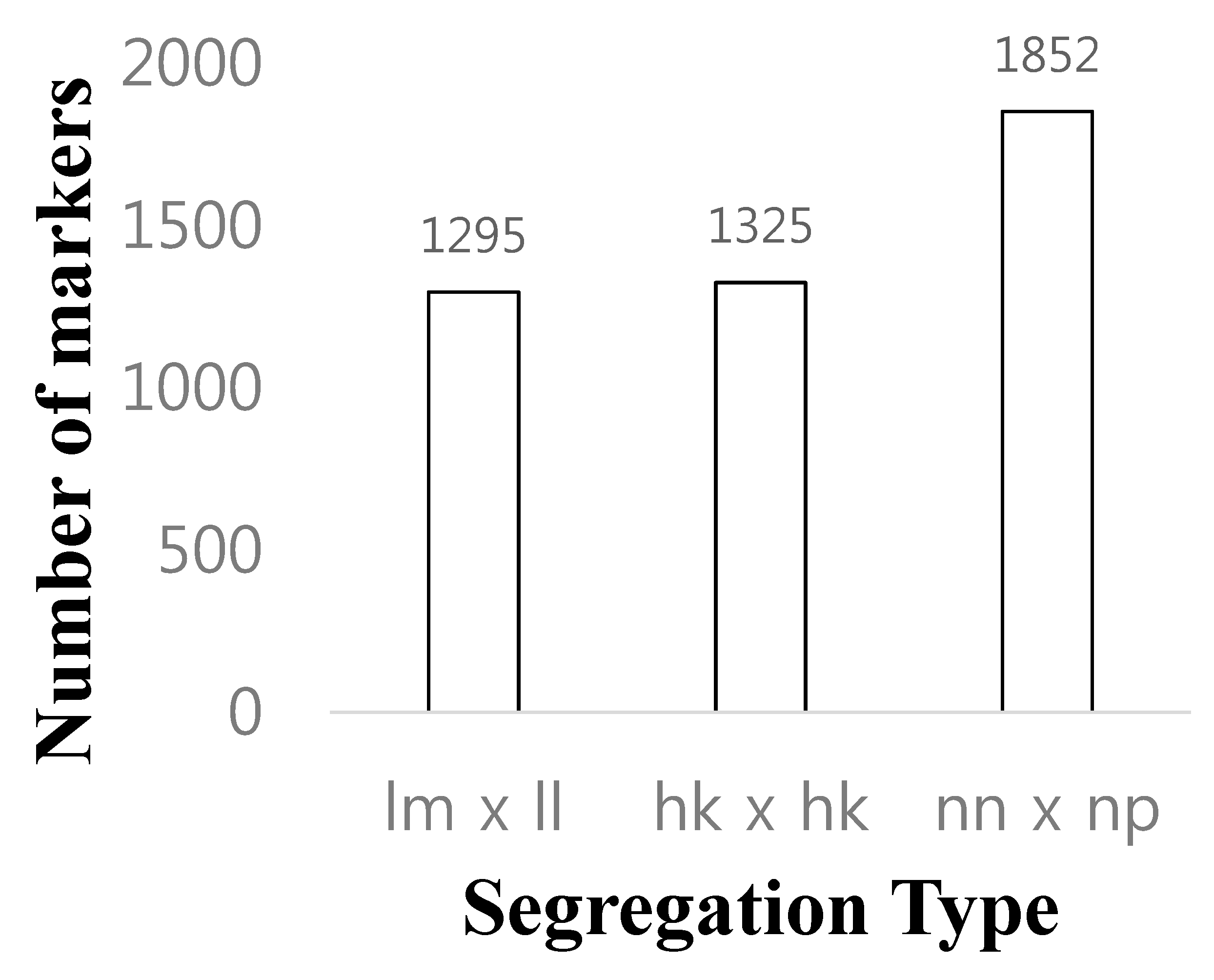
3.1. GBS Library Sequencing and SNP Calling
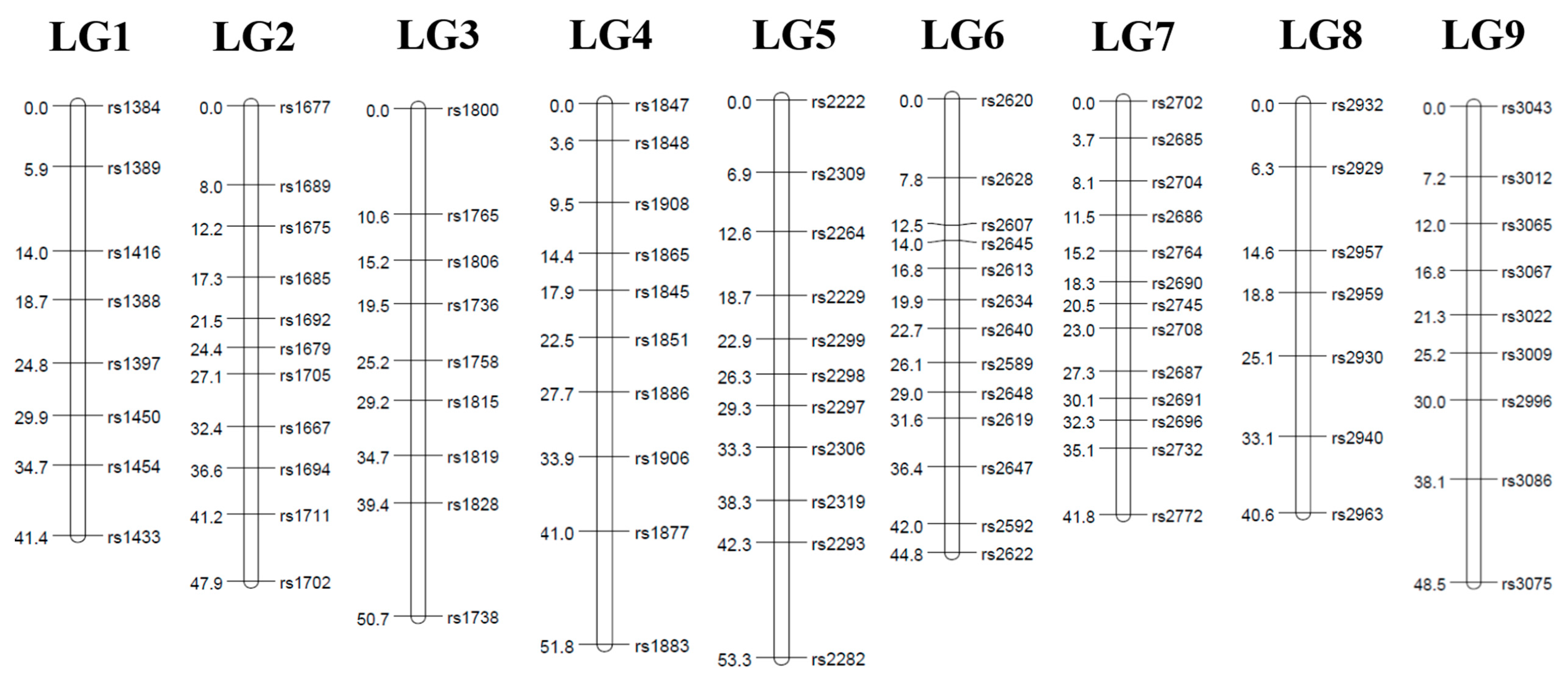
3.2. Linkage Map
3.3. Regeneration Rate from Microspore Culture
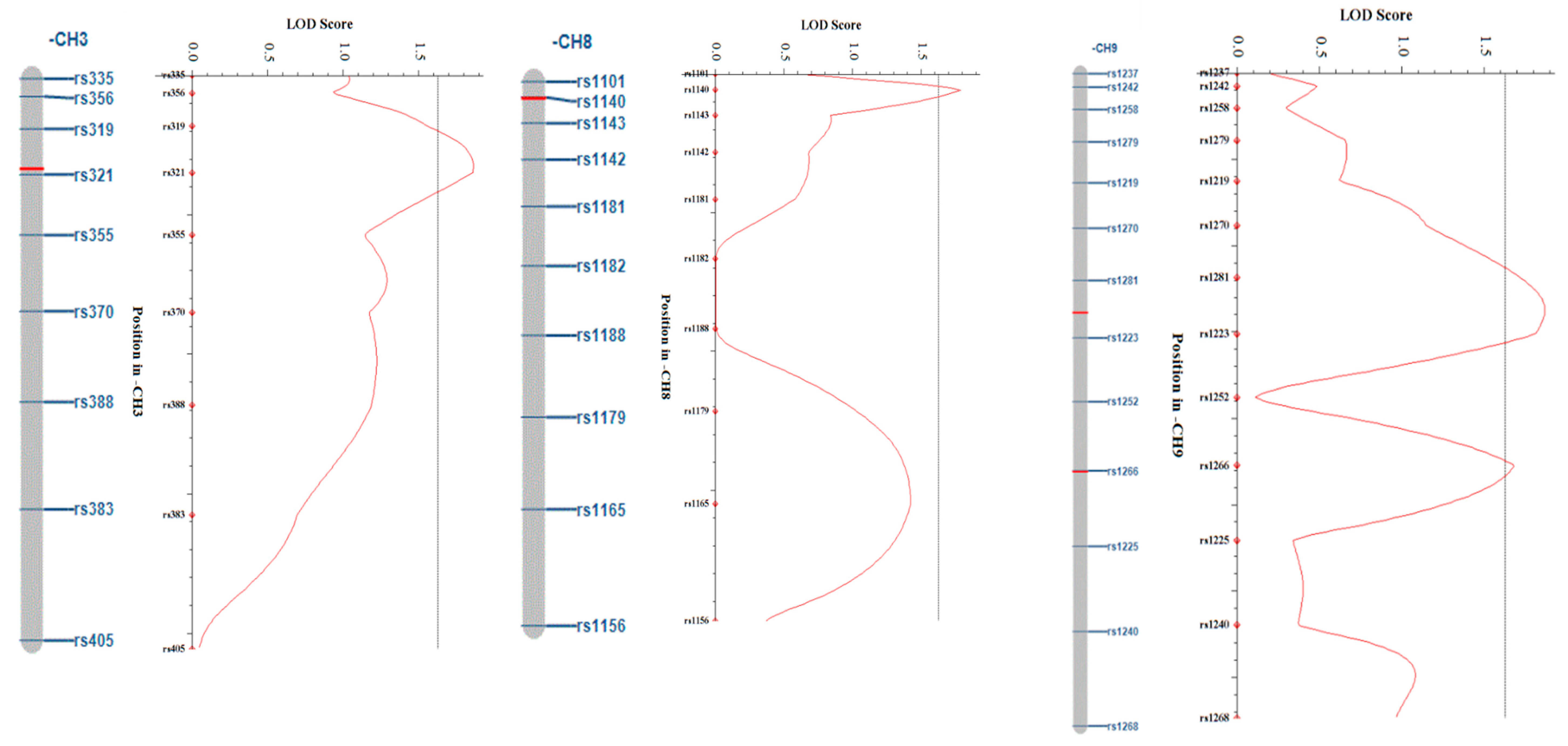
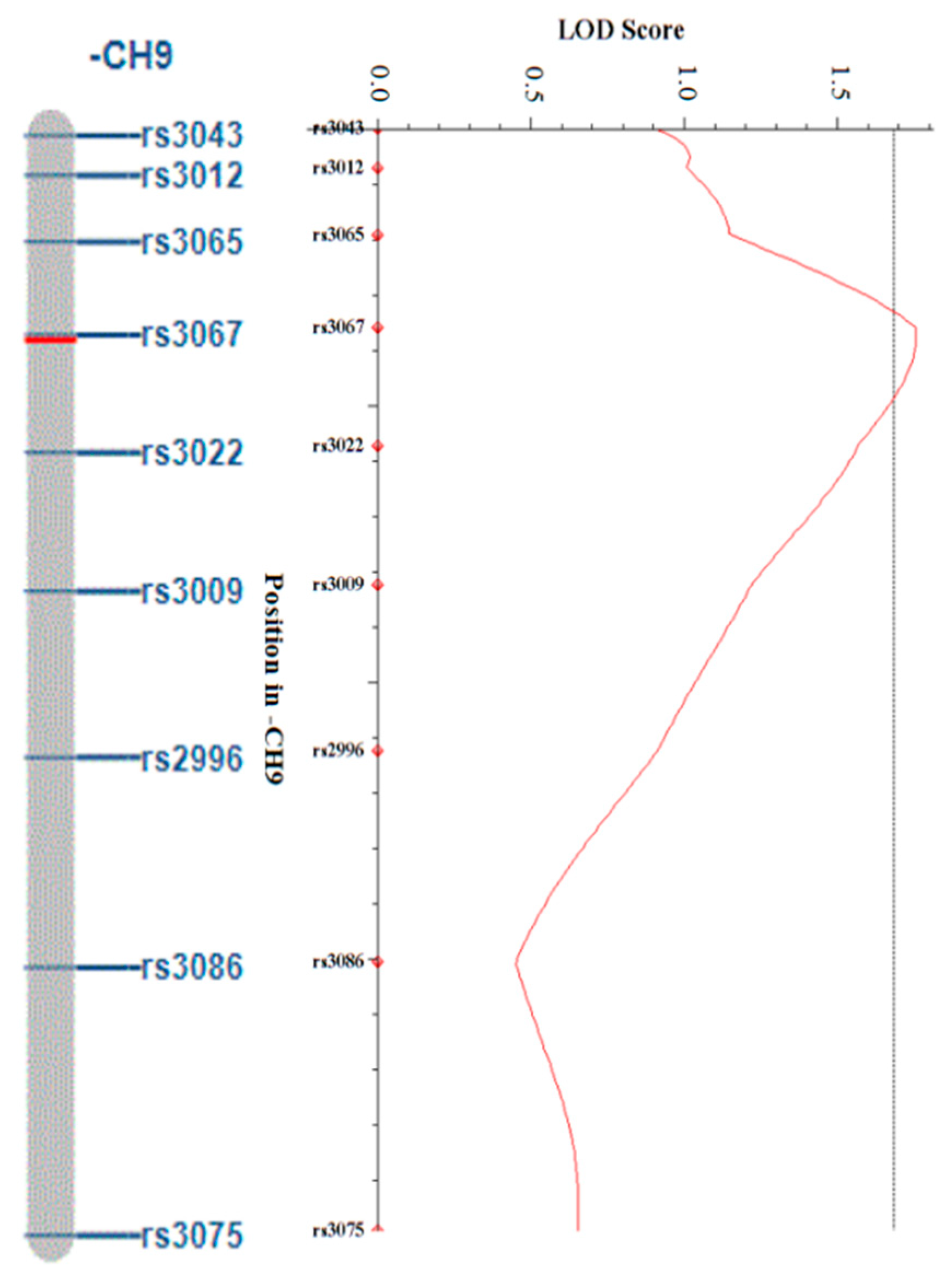
3.4. QTL Mapping Analysis of Regeneration Rate and the Candidate Genes Related to Microspore Culture
4. Discussion
4.1. Phenotypic Variation of Regeneration Rate in Parent Lines and F1 Population
4.2. GBS Analysis Using an NGS Platform and Genetic Mapping Using SNP Markers
4.3. QTLs Associated with Regeneration Rate
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hall, A.E.; Fiebig, A.; Preuss, D. Beyond the Arabidopsis genome: Opportunities for comparative genomics. Plant Physiol. 2002, 129, 1439–1447. [Google Scholar] [CrossRef] [Green Version]
- Schranz, M.E.; Lysak, M.A.; Mitchell-Olds, T. The ABC’s of comparative genomics in the Brassicaceae: Building blocks of crucifer genomes. Trends Plant Sci. 2006, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Moghe, G.D.; Hufnagel, D.E.; Tang, H.; Xiao, Y.; Dworkin, I.; Town, C.D.; Conner, J.K.; Shiu, S.-H. Consequences of whole-genome triplication as revealed by comparative genomic analyses of the wild radish Raphanus raphanistrum and three other Brassicaceae species. Plant Cell 2014, 26, 1925–1937. [Google Scholar] [CrossRef] [Green Version]
- Marie, D.; Brown, S.C. A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biol. Cell 1993, 78, 41–51. [Google Scholar] [CrossRef]
- Johnston, J.S.; Pepper, A.E.; Hall, A.E.; Chen, Z.J.; Hodnett, G.; Drabek, J.; Lopez, R.; Price, H.J. Evolution of genome size in Brassicaceae. Ann. Bot. 2005, 95, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Kitashiba, H.; Li, F.; Hirakawa, H.; Kawanabe, T.; Zou, Z.; Hasegawa, Y.; Tonosaki, K.; Shirasawa, S.; Fukushima, A.; Yokoi, S. Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 2014, 21, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun, J.-H.; Chung, H.; Chung, W.-H.; Oh, M.; Jeong, Y.-M.; Kim, N.; Ahn, B.O.; Park, B.-S.; Park, S.; Lim, K.-B. Construction of a reference genetic map of Raphanus sativus based on genotyping by whole-genome resequencing. Theor. Appl. Genet. 2015, 128, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.H.; Hill, C.B. Rapid-cycling populations of Brassica. Science 1986, 232, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Pink, D.; Bailey, L.; McClement, S.; Hand, P.; Mathas, E.; Buchanan-Wollaston, V.; Astley, D.; King, G.; Teakle, G. Double haploids, markers and QTL analysis in vegetable brassicas. Euphytica 2008, 164, 509–514. [Google Scholar] [CrossRef]
- Kakizaki, Y.; Kasai, T. Bud Pollination in Cabbage and Radish: Some Examples of Conspicuous “Pseudo-fertility” in Normally Self-incompatible Plants. J. Hered. 1933, 24, 359–360. [Google Scholar] [CrossRef]
- Sakamoto, K.; Kusaba, M.; Nishio, T. Single-seed PCR-RFLP analysis for the identification of S haplotypes in commercial F 1 hybrid cultivars of broccoli and cabbage. Plant Cell Rep. 2000, 19, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, T. Studies on the behavior of incompatible pollen in the Japanese radish. J. Jpn. Soc. Hortic. Sci. 1940, 11, 207–234. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Tripathi, S.; Somani, K. Hybrid seed production of radish (Raphanus sativus L.). J. New Seeds 2001, 3, 51–58. [Google Scholar] [CrossRef]
- Cao, M.Q.; Li, Y.; Liu, F.; Doré, C. Embryogenesis and plant regeneration of pakchoi (Brassica rapa L. ssp. chinensis) via in vitro isolated microspore culture. Plant Cell Rep. 1994, 13, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Ham, I.; Park, M.; Kim, T.; Lim, Y.; Lee, E. Seed production ability of doubled haploid plants through microspore culture in Chinese cabbage (Brassica rapa L. ssp. pekinensis) introduced from China. Korean J. Hortic. Sci. Technol. 2012, 30, 573–578. [Google Scholar] [CrossRef]
- Germana, M.A. Gametic embryogenesis and haploid technology as valuable support to plant breeding. Plant Cell Rep. 2011, 30, 839–857. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D. Isolation and quantification of DNA. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; pp. 1–80. [Google Scholar]
- Na, H.; Park, S.; Hwang, G.; Yoon, M.; Chun, C. Medium, AgNO3, activated charcoal and NAA effects on microspore culture in Brassica rapa. Korean J. Hortic. Sci. Technol. 2009, 27, 657–661. [Google Scholar]
- Nitsch, C.; Nitsch, J. The induction of flowering in vitro in stem segments of Plumbago indica L. Planta 1967, 72, 355–370. [Google Scholar] [CrossRef]
- Lichter, R. Induction of haploid plants from isolated pollen of Brassica napus. Z. Pflanzenphysiol. 1982, 105, 427–434. [Google Scholar] [CrossRef]
- Lee, S.; Nam, S. Microspore culture of broccoli (Brassica oleracea var. italica). J. Korean Soc. Hortic. Sci. 1995, 36, 635–640. [Google Scholar]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Chung, Y.S.; Lee, Y.G.; Silva, R.R.; Park, S.; Park, M.Y.; Lim, Y.P.; Choi, S.C.; Kim, C. Potential SNPs related to microspore culture in Raphanus sativus based on a single-marker analysis. Can. J. Plant Sci. 2018, 98, 1072–1083. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.-M.; Kim, N.; Ahn, B.O.; Oh, M.; Chung, W.-H.; Chung, H.; Jeong, S.; Lim, K.-B.; Hwang, Y.-J.; Kim, G.-B. Elucidating the triplicated ancestral genome structure of radish based on chromosome-level comparison with the Brassica genomes. Theor. Appl. Genet. 2016, 129, 1357–1372. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van Oojien, J.; Van Ooijen, J.; Van Ooijen, J.; Van Ooijen, J.; Kyazmay, B.; Ooijen, J.; Riel, J.; van OOIJEN, J.; Camp, N.; van’t Verlaat, J. JoinMap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Population; ScienceOpen, Inc.: Boston, MA, USA, 2006. [Google Scholar]
- Grattapaglia, D.; Sederoff, R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: Mapping strategy and RAPD markers. Genetics 1994, 137, 1121–1137. [Google Scholar]
- Kosambi, D.D. The estimation of map distances from recombination values. In DD Kosambi; Springer: New Delhi, India, 2016; pp. 125–130. [Google Scholar]
- Voorrips, R. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Hashida, T.; Nakatsuji, R.; Budahn, H.; Schrader, O.; Peterka, H.; Fujimura, T.; Kubo, N.; Hirai, M. Construction of a chromosome-assigned, sequence-tagged linkage map for the radish, Raphanus sativus L. and QTL analysis of morphological traits. Breed. Sci. 2013, 63, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Choi, S.R.; Dhandapani, V.; Rameneni, J.J.; Li, X.; Pang, W.; Lee, J.-Y.; Lim, Y.P. Quantitative trait loci for morphological traits and their association with functional genes in Raphanus sativus. Front. Plant Sci. 2016, 7, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.E.; Keller, W.A.; Arnison, P.G. Experimental haploidy in Brassica species. In In Vitro Haploid Production in Higher Plants; Springer: Dordrecht, The Netherlands, 1996; pp. 143–172. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, S.M.; Jaganathan, D.; Ruperao, P.; Chen, C.; Punna, R.; Kudapa, H.; Thudi, M.; Roorkiwal, M.; Katta, M.A.; Doddamani, D. Prioritization of candidate genes in “QTL-hotspot” region for drought tolerance in chickpea (Cicer arietinum L.). Sci. Rep. 2015, 5, 15296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Wang, L.; Gong, Y.; Dai, W.; Wang, Y.; Zhu, X.; Wen, T.; Liu, L. Genetic linkage map construction and QTL mapping of cadmium accumulation in radish (Raphanus sativus L.). Theor. Appl. Genet. 2012, 125, 659–670. [Google Scholar] [CrossRef]
- Zou, Z.; Ishida, M.; Li, F.; Kakizaki, T.; Suzuki, S.; Kitashiba, H.; Nishio, T. QTL analysis using SNP markers developed by next-generation sequencing for identification of candidate genes controlling 4-methylthio-3-butenyl glucosinolate contents in roots of radish, Raphanus sativus L. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Firoozabady, E.; DeBoer, D. Plant regeneration via somatic embryogenesis in many cultivars of cotton (Gossypium hirsutum L.). Vitr. Cell. Dev. Boil. Anim. 1993, 29, 166–173. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Abe, T.; Sasahara, T. RFLP mapping of QTLs influencing shoot regeneration from mature seed-derived calli in rice. Crop Sci. 2000, 40, 245–247. [Google Scholar] [CrossRef]
- Kwon, Y.-S.; Kim, K.-M.; Eun, M.-Y.; Sohn, J.-K. Quantitative trait loci mapping associated with plant regeneration ability from seed derived calli in rice (Oryza sativa L.). Mol. Cells 2001, 11, 64–67. [Google Scholar]
- Ma, J.; Deng, M.; Lv, S.-Y.; Yang, Q.; Jiang, Q.-T.; Qi, P.-F.; Li, W.; Chen, G.-Y.; Lan, X.-J.; Wei, Y.-M. Identification of QTLs associated with tissue culture response of mature wheat embryos. SpringerPlus 2016, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Komatsuda, T.; Annaka, T.; Oka, S. Genetic mapping of a quantitative trait locus (QTL) that enhances the shoot differentiation rate in Hordeum vulgare L. Theor. Appl. Genet. 1993, 86, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Takahashi, H.; Sato, K.; Takeda, K. Mapping genes for callus growth and shoot regeneration in barley (Hordeum vulgare L.). Jpn. J. Breed. 1996, 46, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Bregitzer, P.; Campbell, R.D. Genetic markers associated with green and albino plant regeneration from embryogenic barley callus. Crop Sci. 2001, 41, 173–179. [Google Scholar] [CrossRef]
- Holme, I.; Torp, A.; Hansen, L.; Andersen, S. Quantitative trait loci affecting plant regeneration from protoplasts of Brassica oleracea. Theor. Appl. Genet. 2004, 108, 1513–1520. [Google Scholar] [CrossRef]
- Torp, A.; Hansen, A.; Andersen, S.B. Chromosomal regions associated with green plant regeneration in wheat (Triticum aestivum L.) anther culture. Euphytica 2001, 119, 377. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, K.; Eun, M.; Sohn, J. QTL mapping and associated marker selection for the efficacy of green plant regeneration in anther culture of rice. Plant Breed. 2002, 121, 10–16. [Google Scholar] [CrossRef]
- Mano, Y.; Komatsuda, T. Identification of QTLs controlling tissue-culture traits in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2002, 105, 708–715. [Google Scholar] [CrossRef]
- Bolibok, H.; Rakoczy-Trojanowska, M. Genetic mapping of QTLs for tissue-culture response in plants. Euphytica 2006, 149, 73–83. [Google Scholar] [CrossRef]
- Jia, H.; Yi, D.; Yu, J.; Xue, S.; Xiang, Y.; Zhang, C.; Zhang, Z.; Zhang, L.; Ma, Z. Mapping QTLs for tissue culture response of mature wheat embryos. Mol. Cells 2007, 23, 323–330. [Google Scholar]
- Tyagi, N.; Dahleen, L.S.; Bregitzer, P. Candidate genes within tissue culture regeneration QTL revisited with a linkage map based on transcript-derived markers. Crop Sci. 2010, 50, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Duijs, J.; Voorrips, R.; Visser, D.; Custers, J. Microspore culture is successful in most crop types of Brassica oleracea L. Euphytica 1992, 60, 45–55. [Google Scholar]
- Custers, J.B.; Cordewener, J.H.; Nöllen, Y.; Dons, H.J.; Campagne, M.M.V.L. Temperature controls both gametophytic and sporophytic development in microspore cultures of Brassica napus. Plant Cell Rep. 1994, 13, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Möllers, C.; Röbbelen, G. Increased embryogenesis after colchicine treatment of microspore cultures of Brassica napus L. J. Plant Physiol. 1994, 143, 222–226. [Google Scholar] [CrossRef]
- Hansen, N.; Andersen, S.B. In vitro chromosome doubling potential of colchicine, oryzalin, trifluralin, and APM in Brassica napus microspore culture. Euphytica 1996, 88, 159–164. [Google Scholar] [CrossRef]
- Sato, S.; Katoh, N.; Iwai, S.; Hagimori, M. Effect of low temperature pretreatment of buds or inflorescence on isolated microspore culture in Brassica rapa (syn. B. campestris). Breed. Sci. 2002, 52, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Chun, C.; Park, H.; Na, H. Microspore-derived embryo formation in radish (Raphanus sativus L.) according to nutritional and environmental conditions. Hortic. Environ. Biotechnol. 2011, 52, 530. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Favero, D.S.; Sakamoto, Y.; Iwase, A.; Coleman, D.; Rymen, B.; Sugimoto, K. Molecular mechanisms of plant regeneration. Annu. Rev. Plant Biol. 2019, 70, 377–406. [Google Scholar] [CrossRef]
- Zhao, Y.; Christensen, S.K.; Fankhauser, C.; Cashman, J.R.; Cohen, J.D.; Weigel, D.; Chory, J. A role for flavin monooxygenase-like enzymes in auxin biosynthesis. Science 2001, 291, 306–309. [Google Scholar] [CrossRef]
- Liu, J.; Sheng, L.; Xu, Y.; Li, J.; Yang, Z.; Huang, H.; Xu, L. WOX11 and 12 are involved in the first-step cell fate transition during de novo root organogenesis in Arabidopsis. Plant Cell 2014, 26, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Tong, J.; Xiao, L.; Ruan, Y.; Liu, J.; Zeng, M.; Huang, H.; Wang, J.-W.; Xu, L. YUCCA-mediated auxin biogenesis is required for cell fate transition occurring during de novo root organogenesis in Arabidopsis. J. Exp. Bot. 2016, 67, 4273–4284. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.J.; Wang, L.; Sun, W.; Zhang, Y.; Zhou, C.; Su, Y.H.; Li, W.; Sun, T.T.; Zhao, X.Y.; Li, X.G. Pattern of auxin and cytokinin responses for shoot meristem induction results from the regulation of cytokinin biosynthesis by AUXIN RESPONSE FACTOR3. Plant Physiol. 2013, 161, 240–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Harashima, H.; Shibata, M.; Ohnuma, M.; Breuer, C.; Morao, A.K.; de Lucas, M.; De Veylder, L. PRC2 represses dedifferentiation of mature somatic cells in Arabidopsis. Nat. Plants 2015, 1, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozgová, I.; Muñoz-Viana, R.; Hennig, L. PRC2 represses hormone-induced somatic embryogenesis in vegetative tissue of Arabidopsis thaliana. PLoS Genet. 2017, 13, e1006562. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LGs | Length (cM) | Number of SNPs | Average Interval (cM) | Largest Interval (cM) |
|---|---|---|---|---|
| 1 | 40.2 | 7 | 6.7 | 8.8 |
| 2 | 36.7 | 7 | 6.1 | 7.9 |
| 3 | 47.9 | 9 | 6.0 | 8.5 |
| 4 | 55.2 | 10 | 6.1 | 10.8 |
| 5 | 56.0 | 13 | 4.7 | 13.4 |
| 6 | 55.2 | 11 | 5.5 | 10.9 |
| 7 | 51.7 | 7 | 8.6 | 11.8 |
| 8 | 42.2 | 10 | 4.7 | 8.9 |
| 9 | 54.1 | 13 | 4.5 | 7.7 |
| Total | 439.2 | 87 | 5.9 |
| LGs | Length (cM) | Number of SNPs | Average Interval (cM) | Largest Interval (cM) |
|---|---|---|---|---|
| 1 | 41.4 | 8 | 5.9 | 3.1 |
| 2 | 47.9 | 11 | 4.8 | 6.7 |
| 3 | 50.7 | 9 | 6.3 | 11.3 |
| 4 | 51.8 | 10 | 5.8 | 10.8 |
| 5 | 53.3 | 11 | 4.2 | 11 |
| 6 | 44.8 | 13 | 3.7 | 7.8 |
| 7 | 41.8 | 13 | 3.5 | 6.7 |
| 8 | 40.6 | 7 | 6.8 | 8.3 |
| 9 | 48.5 | 9 | 6.0 | 10.4 |
| Total | 420.8 | 91 | 5.2 |
| F1 Individuals | |
|---|---|
| Total embryo average 1 | 3.52 3 |
| Total culture average 2 | 1.71 |
| QTL | LOD | A.E1 | PVE(%) 2 |
|---|---|---|---|
| LG3_1 | 1.86 | −6.13 | 5.81 |
| LG8_1 | 1.79 | −5.96 | 5.54 |
| LG9_1 | 1.87 | −6.38 | 6.25 |
| LG9_2 | 1.68 | −5.74 | 5.24 |
| QTL | LOD | A.E 1 | PVE(%) 2 |
|---|---|---|---|
| LG9_1 | 1.75 | 5.90 | 13.19 |
| QTL | Gene ID | A.T ortholog | Gene Description |
|---|---|---|---|
| P1_Chr8_11 | Rs426380 | AT4G32540 | Flavin-binding monooxygenase family protein |
| P1_Chr8_1 | Rs426400 | AT4G32540 | Flavin-binding monooxygenase family protein |
| P1_Chr9_1/P1_Chr9_2 | Rs465100 | AT5G51230 | VEFS-Box of polycomb protein |
| P2_Chr9_12 | Rs479580 | AT4G02020 | SET domain-containing protein |
| P2_Chr9_1 | Rs479680 | AT4G02020 | SET domain-containing protein |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Kang, Y.; Lee, S.-J.; Choi, S.-H.; Jeon, D.-H.; Park, M.-Y.; Park, S.; Lim, Y.P.; Kim, C. Quantitative Trait Loci (QTLs) Associated with Microspore Culture in Raphanus sativus L. (Radish). Genes 2020, 11, 337. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030337
Kim K, Kang Y, Lee S-J, Choi S-H, Jeon D-H, Park M-Y, Park S, Lim YP, Kim C. Quantitative Trait Loci (QTLs) Associated with Microspore Culture in Raphanus sativus L. (Radish). Genes. 2020; 11(3):337. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030337
Chicago/Turabian StyleKim, Kyeongmin, Yuna Kang, Sol-Ji Lee, Se-Hyun Choi, Dong-Hyun Jeon, Min-Young Park, Suhyoung Park, Yong Pyo Lim, and Changsoo Kim. 2020. "Quantitative Trait Loci (QTLs) Associated with Microspore Culture in Raphanus sativus L. (Radish)" Genes 11, no. 3: 337. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030337