A DSG1 Frameshift Variant in a Rottweiler Dog with Footpad Hyperkeratosis
by
 , , and
, , and
Katherine A. Backel
1,†, 
Sarah Kiener
2,3,†,
Vidhya Jagannathan
2,3,
Margret L. Casal
1,
Tosso Leeb
2,3,*,‡ and
Elizabeth A. Mauldin
1,‡ 1
School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
2
Institute of Genetics, Vetsuisse Faculty, University of Bern, 3001 Bern, Switzerland
3
Dermfocus, University of Bern, 3001 Bern, Switzerland
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work (shared first authors).
‡
These authors contributed equally to this work (shared senior authors).
Genes 2020, 11(4), 469; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040469
Submission received: 1 April 2020
/
Revised: 23 April 2020
/
Accepted: 23 April 2020
/
Published: 24 April 2020
(This article belongs to the Special Issue Molecular Basis of Inherited Diseases in Companion Animals)
Abstract
:A single male Rottweiler dog with severe footpad hyperkeratosis starting at an age of eight weeks was investigated. The hyperkeratosis was initially restricted to the footpads. The footpad lesions caused severe discomfort to the dog and had to be trimmed under anesthesia every 8–10 weeks. Histologically, the epidermis showed papillated villous projections of dense keratin in the stratum corneum. Starting at eight months of age, the patient additionally developed signs consistent with atopic dermatitis and recurrent bacterial skin and ear infections. Crusted hyperkeratotic plaques developed at sites of infection. We sequenced the genome of the affected dog and compared the data to 655 control genomes. A search for variants in 32 candidate genes associated with human palmoplantar keratoderma (PPK) revealed a single private protein-changing variant in the affected dog. This was located in the DSG1 gene encoding desmoglein 1. Heterozygous monoallelic DSG1 variants have been reported in human patients with striate palmoplantar keratoderma I (SPPK1), while biallelic DSG1 loss of function variants in humans lead to a more pronounced condition termed severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome. The identified canine variant, DSG1:c.2541_2545delGGGCT, leads to a frameshift and truncates about 20% of the coding sequence. The affected dog was homozygous for the mutant allele. The comparative data on desmoglein 1 function in humans suggest that the identified DSG1 variant may have caused the footpad hyperkeratosis and predisposition for allergies and skin infections in the affected dog.
1. Introduction
The skin forms an essential barrier against the environment. In humans, the soles of the feet and the palms of the hands are covered by the specially structured palmoplantar epidermis, which has to bear the strongest mechanical forces of the entire skin. Genodermatoses characterized by altered structural and junctional proteins of these specialized regions comprise the palmoplantar keratodermas (PPK), a diverse group of inherited disorders collectively characterized by excessive or abnormal thickening of the palmoplantar skin. Variants in at least 32 genes have been shown to cause different forms of isolated or syndromic PPK in humans (Table 1) [1,2].
Footpad hyperkeratosis in dogs is a genetically heterogenous group of inherited diseases corresponding to human PPK. So far, causative genetic variants for two different forms of canine footpad hyperkeratosis have been reported. Hereditary footpad hyperkeratosis (HFH) in Irish Terriers and Kromfohrländer dogs is caused by a variant in the FAM83G gene [36] encoding a protein involved in BMP and WNT signaling [37,38,39]. The syndromic HFH phenotype is characterized by an orthokeratotic hyperplasia of the footpad epidermis in combination with an irregular hair morphology. FAM83G variants have also been described in human patients with PPK and exuberant scalp hair [12] and the wooly mouse mutant [40]. A KRT16 frameshift variant has been reported in Dogues de Bordeaux with focal nonepidermolytic footpad hyperkatosis [41]. Interestingly, this disease is inherited as an autosomal recessive trait, while the human forms of KRT16-associated focal nonepidermolytic PPK typically are inherited as autosomal dominant traits [25,26].
This study was initiated after an owner reported a juvenile male Rottweiler dog suffering from footpad hyperkeratosis. The goal of the study was to characterize the clinical and histopathological phenotype and to identify a possible underlying causative genetic defect.
2. Materials and Methods
2.1. Ethics Statement
All animal experiments were performed according to local regulations. The dog in this study is privately owned and was examined with the consent of the owner. The "Cantonal Committee for Animal Experiments" approved the collection of blood samples (Canton of Bern; permit 75/16).
2.2. Animal Selection
A male Rottweiler dog with footpad hyperkeratosis was investigated. Footpad biopsies were collected at initial presentation to rule out infectious and inflammatory causes of hyperkeratosis. The clinical presentation was inconsistent with other causes of secondary hyperkeratosis. An EDTA blood sample was collected for genomic DNA isolation. Additionally, we used 15 blood samples from other Rottweilers, which had been donated to the Vetsuisse Biobank. They represented population controls without reports of footpad hyperkeratosis. The photo of the control Rottweiler, shown in Figure 1B, represents a stock photo from the University of Pennsylvania (UPENN) veterinary hospital. This dog was not genotyped. The biopsy of the control dog, shown in Figure 2A, originates from a six-month-old healthy Beagle that was part of another IACUC-approved study at the UPENN School of Veterinary Medicine.
2.3. Histopathological Examinations
Two 6 mm punch biopsies from the footpads were obtained under general anesthesia. The samples were fixed in 10% neutral buffered formalin and routinely processed, including staining with hematoxylin and eosin.
2.4. DNA Extraction
Genomic DNA was isolated from EDTA blood with the Maxwell RSC Whole Blood Kit using a Maxwell RSC instrument (Promega, Dübendorf, Switzerland).
2.5. Whole-Genome Sequencing
An Illumina TruSeq PCR-free DNA library with ~500 bp insert size of the affected dog (RO015) was prepared. We collected 329 million 2 × 150 bp paired-end reads on a NovaSeq 6000 instrument (37x coverage). Mapping and alignment were performed as described [42]. The sequence data were deposited under the study accession PRJEB16012 and the sample accession SAMEA6249501 at the European Nucleotide Archive.
2.6. Variant Calling
Variant calling was performed using GATK HaplotypeCaller [43] in gVCF mode as described [42]. To predict the functional effects of the called variants, SnpEff [44] software together with NCBI annotation release 105 for the CanFam3.1 genome reference assembly was used. For variant filtering we used 655 control genomes (Table S1).
2.7. Gene Analysis
We used the CanFam3.1 dog reference genome assembly and NCBI annotation release 105. Numbering within the canine DSG1 gene corresponds to the NCBI RefSeq accession numbers NM_001002939.1 (mRNA) and NP_001002939.1 (protein).
2.8. Sanger Sequencing
The DSG1:c.2541_2545delGGGCT variant was genotyped by direct Sanger sequencing of PCR amplicons. A 402 bp (or 397 bp in case of the mutant allele) PCR product was amplified from genomic DNA using AmpliTaqGold360Mastermix (Thermo Fisher Scientific, Waltham, MA, USA) together with primers 5‘-GAG CAC TGA ACC GAT TTG CC -3‘ (Primer F) and 5’- GGC ATA GTC AAA GAG GTG GGT-3’ (Primer R). After treatment with exonuclease I and alkaline phosphatase, amplicons were sequenced on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific). Sanger sequences were analyzed using the Sequencher 5.1 software (GeneCodes, Ann Arbor, MI, USA).
3. Results
3.1. Clinical Examination
A six-month-old male intact Rottweiler dog presented for evaluation of thick, rapidly growing footpads and discomfort (shifting stance, unwilling to stand for long periods). An unusual appearance of the pads (described as “dryness”) had been first noted by the owners when the dog was obtained at eight weeks of age. At the time of the initial presentation, the patient was otherwise healthy with no other systemic or dermatologic signs. On examination, all pads on all feet were markedly thickened by dense mounds of adherent keratin (Figure 1). The digital pads and metatarsal/metacarpal pads were the most severely affected. Fissures and mobile keratin ridges were present along the edges of the pads. There was mild diffuse scale over the trunk, which was considered to be within normal limits.
Regular application of moisturizers (urea), keratolytic (propylene glycol), and keratoplastic agents (salicylic acid/sulfur) and regular home trimming was recommended. The patient was unable to tolerate oral retinoids (isotretinoin). Initially, physical trimming of footpads under general anesthesia was performed every 4–6 months. However, the frequency by which this was required increased over time, and by the third year of life, it was performed every 8–10 weeks. To address significant discomfort, the patient was also started on gabapentin and codeine for pain control.
Additionally, between 8–12 months of age, the patient developed mild nonseasonal pruritus and recurrent ear infections. The patient was placed on an isoxazoline (Bravecto®) for parasite control. A 10-week strict hydrolyzed protein (Royal Canin Ultamino®) diet trial was performed without improvement, and the patient was presumptively diagnosed with atopic dermatitis. At one year of age, the patient was started on 0.5 mg/kg Oclacitinib (Apoquel®) for control of pruritus.
Despite control of pruritus, the patient continued to develop recurrent ear infections and intermittent episodes of bacterial skin infection. Superficial bacterial infections developed most frequently in areas of heavy wear (elbows, lateral hocks) but were also found occasionally on the trunk, neck, and glabrous areas. At sites of infection, hyperkeratotic plaques with superficial crusting were noted (Figure S1). Infections were managed with topical antiseptics and systemic culture-based antibiotics when indicated.
3.2. Histopathological Findings
Histologically, the stratum corneum was markedly expanded by villous projections of orthokeratotic hyperkeratosis. The subtending granular layer of the epidermis was mildly hyperplastic. The samples had no significant inflammation (Figure 2).
3.3. Genetic Analysis
We sequenced the genome of the affected dog and searched for homozygous and heterozygous variants in 32 known candidate genes (Table 1) that were not present in the genome sequences of 647 control dogs and 8 wolves (Table 2, Table S2).
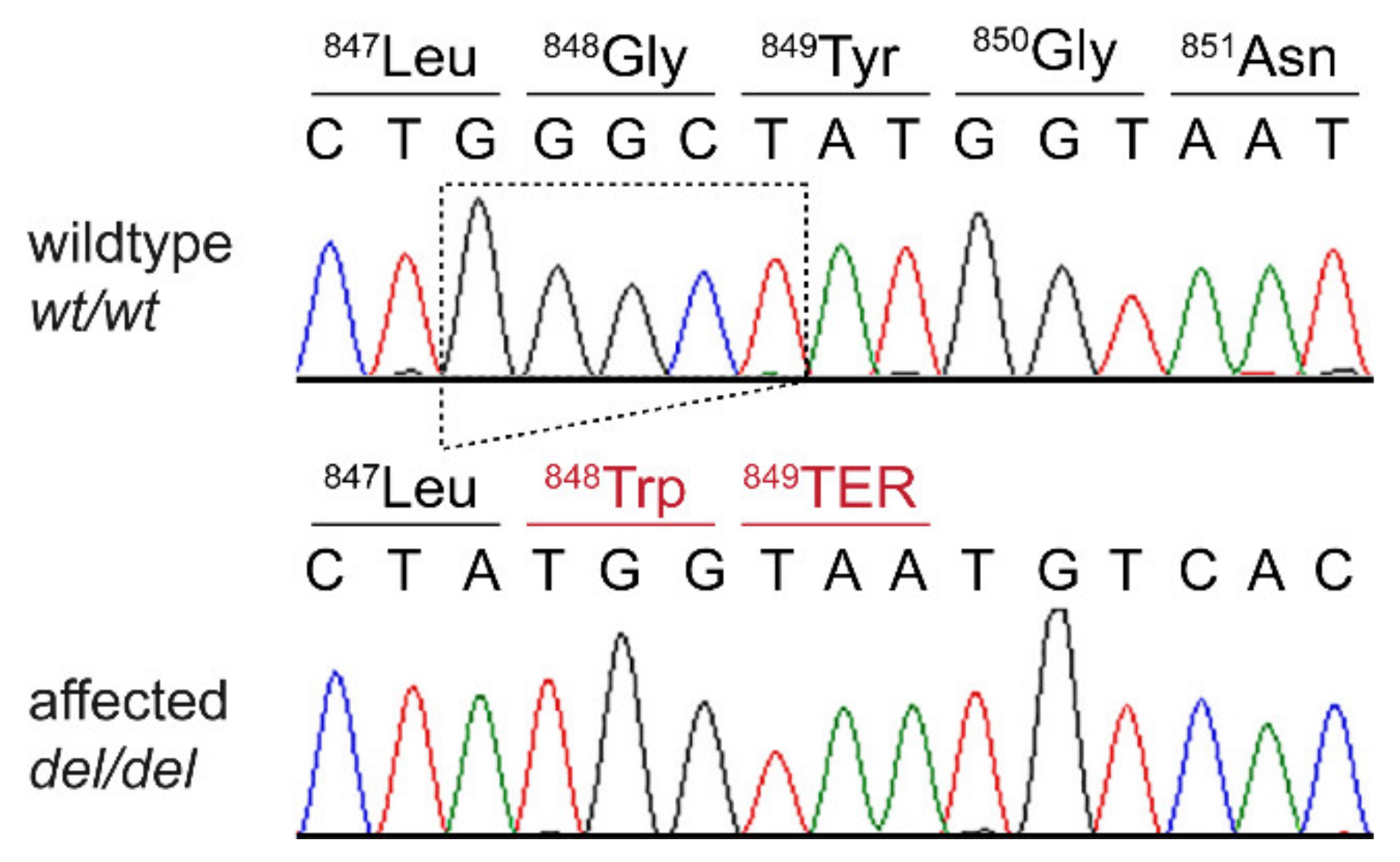
This analysis identified a single homozygous private protein-changing variant in DSG1, a known candidate gene for palmoplantar keratoderma in humans [9]. The variant, a 5 bp deletion, can be designated as Chr7:58,163,636_58,163,640del5 (CanFam3.1 assembly). It is a frameshift variant, NM_001002939.1:c.2541_2545delGGGCT, predicted to truncate 207 amino acids from the C-terminus of the wildtype DSG1 protein, NP_001002939.1:p.(Gly848Trpfs*2). We did not investigate whether any mutant protein is expressed or whether the premature stop codon caused by the frameshift deletion leads to nonsense-mediated decay of the transcript.
We confirmed the presence of the 5 bp coding deletion in DSG1 by Sanger sequencing and genotyped 15 control Rottweiler dogs. The case was homozygous for the mutant allele, while none of the 15 control dogs carried this allele (Figure 3).
4. Discussion
In this study, we identified a homozygous DSG1:c.2541_2545delGGGCT frameshift variant in a Rottweiler dog with severe footpad hyperkeratosis. DSG1 encodes desmoglein 1, a calcium-binding transmembrane glycoprotein of the cadherin family. Desmoglein 1 represents a major component of desmosomes that mediates cell–cell adhesion between keratinocytes in the upper layers of the epidermis [45]. Intact desmosomes are essential to maintain the skin barrier function [45]. Desmoglein 1 also represents the major autoantigen in human pemphigus foliaceus [46].
Variants in human DSG1 cause striate palmoplantar keratoderma I (SPPK1). It is interesting to note that SPKK1 in humans is an autosomal dominant phenotype with the pathogenic variants being present in a heterozygous state in affected individuals [9,47]. In humans, SPKK1 is caused by haploinsufficiency of desmoglein 1 [9,47]. The investigated dog of our study was homozygous for a presumed null allele of DSG1 and unlikely to express any functional desmoglein 1. In humans, a rare syndromic form of PPK referred to as SAM syndrome has been reported in patients with biallelic DSG1 loss-of-function variants [48]. Subsequent studies of further human patients with biallelic DSG1 variants confirmed the dermatitis and multiple allergies but failed to replicate the reported malabsorption and metabolic wasting [49,50]. The clinical presentation of the affected dog in our study, including the development of atopic dermatitis and hyperkeratotic lesions at sites of bacterial skin infection, resembles the phenotype of human SAM syndrome patients without the metabolic wasting, similar to what has been reported in several human cases [49,50].
The histopathological alterations in the footpad skin of the affected Rottweiler were comparable to the changes seen in footpad hyperkeratosis of FAM83G mutant Irish Terriers and Kromfohrländer dogs [36] and KRT16 mutant Dogues de Bordeaux [41]. Thus, the histopathology cannot predict the specific underlying genetic defect. The clinical phenotype of the studied Rottweiler was more severe than in the previously described canine inherited footpad hyperkeratoses [36,41] and required periodical trimming of the excessively hyperkeratotic footpads. In addition to the severely affected footpads, the DSG1 mutant Rottweiler also had an allergic skin disease and was prone to repeated bacterial skin infections. Such features have not been reported in FAM83G mutant Irish Terriers or Kromfohrländer dogs [36] or in KRT16 mutant Dogues de Bordeaux [41].
Unfortunately, we did not have access to the parents of the affected dog or any other heterozygous dog. It would be interesting to investigate whether heterozygous dogs have completely normal footpads or whether they exhibit a mild phenotype that might go unnoticed by their owners.
In summary, we identified a Rottweiler dog with severe footpad hyperkeratosis that clinically and genetically resembled human SAM syndrome without metabolic wasting. To the best of our knowledge, this dog represents the first nonhuman patient with a spontaneous DSG1 gene defect.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4425/11/4/469/s1, Figure S1: Hyperkeratotic plaque at the elbow of the affected dog. Table S1: Accession numbers of 648 dog and 8 wolf genome sequences; Table S2: Private variants in the sequenced Rottweiler with footpad hyperkeratosis.
Author Contributions
Conceptualization, T.L. and E.A.M.; methodology, V.J.; investigation, K.A.B., S.K., V.J., M.L.C., T.L., and E.A.M..; data curation, V.J.; writing—original draft preparation, K.A.B., S.K., T.L., and E.A.M.; writing—review and editing, K.A.B., S.K., V.J., M.L.C., T.L., and E.A.M.; supervision, T.L. and E.A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors are grateful to the dog owner who donated samples and participated in the study. We thank Eva Andrist, Nathalie Besuchet Schmutz, Sabrina Schenk, and Daniela Steiner for expert technical assistance; the Next Generation Sequencing Platform of the University of Bern for performing the high-throughput sequencing experiments; and the Interfaculty Bioinformatics Unit of the University of Bern for providing high-performance computing infrastructure.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lemke, J.R.; Kernland-Lang, K.; Hörtnagel, K.; Itin, P. Monogenic human skin disorders. Dermatology 2014, 229, 55–64. [Google Scholar] [CrossRef]
- Has, C.; Technau-Hafsi, K. Palmoplantar keratodermas: Clinical and genetic aspects. J. Dtsch. Dermatol. Ges. 2016, 14, 123–139. [Google Scholar] [CrossRef]
- Pohler, E.; Mamai, O.; Hirst, J.; Zamiri, M.; Horn, H.; Nomura, T.; Irvine, A.D.; Moran, B.; Wilson, N.J.; Smith, F.J.D.; et al. Haploinsufficiency for AAGAB causes clinically heterogenous forms of punctate palmoplantar keratoderma. Nat. Genet. 2012, 44, 1272–1276. [Google Scholar] [CrossRef] [Green Version]
- Giehl, K.A.; Eckstein, G.N.; Pasternack, S.M.; Praetzel-Wunder, S.; Ruzicka, T.; Lichtner, P.; Seidl, K.; Rogers, M.; Graf, E.; Langbein, L.; et al. Nonsense mutations in AAGAB cause punctate palmoplantar keratoderma type Buschke-Fischer-Brauer. Am. J. Hum. Genet. 2012, 91, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Blaydon, D.C.; Lind, L.K.; Plagnol, V.; Linton, K.J.; Smith, F.J.D.; Wilson, N.J.; McLean, W.H.I.; Munro, C.S.; South, A.P.; Leigh, I.M.; et al. Mutations in AQP5, encoding a water-channel protein, cause autosomal-dominant diffuse nonepidermolytic palmoplantar keratoderma. Am. J. Hum. Genet. 2013, 93, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Fuchs-Telem, D.; Sarig, O.; van Steensel, M.A.M.; Isakov, O.; Israeli, S.; Nousbeck, J.; Richard, K.; Winnepenninckx, V.; Vernooij, M.; Shomron, N.; et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet. 2012, 91, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.-R.; Zhang, X.; Chen, G.; Zhang, J.-G.; Sun, L.-D.; Du, W.; Zhang, Q.; Cui, Y.; Zhu, J.; Tang, X.-F.; et al. Exome sequencing identifies a COL14A1 mutation in a large Chinese pedigree with punctate palmoplantar keratoderma. J. Med. Genet. 2012, 49, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Toomes, C.; James, J.; Wood, A.J.; Wu, C.L.; McCormick, D.; Lench, N.; Hewitt, C.; Moynihan, L.; Roberts, E.; Woods, C.G.; et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat. Genet. 1999, 23, 421–424. [Google Scholar] [CrossRef]
- Rickman, L.; Simrak, D.; Stevens, H.P.; Hunt, D.M.; King, I.A.; Bryant, S.P.; Eady, R.A.J.; Leigh, I.M.; Arnemann, J.; Magee, A.I.; et al. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum. Mol. Genet. 1999, 8, 971–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.K.; McKenna, K.E.; Purkis, P.E.; Green, K.J.; Eady, R.A.J.; Leigh, I.M.; Hughes, A.E. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum. Mol. Genet. 1999, 8, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eytan, O.; Morice-Picard, F.; Sarig, O.; Ezzedine, K.; Isakov, O.; Li, Q.; Ishida-Yamamoto, A.; Shomron, N.; Goldsmith, T.; Fuchs-Telem, D.; et al. Cole disease results from mutations in ENPP1. Am. J. Hum. Genet. 2013, 93, 752–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruthappu, T.; McGinty, L.A.; Blaydon, D.C.; Fell, B.; Määttä, A.; Duit, R.; Hawkins, T.; Braun, K.M.; Simpson, M.A.; O’Toole, E.A.; et al. Recessive mutation in FAM83G associated with palmoplantar keratoderma and exuberant scalp hair. J. Investig. Dermatol. 2018, 138, 984–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cao, X.; Lin, Z.; Lee, M.; Jia, X.; Ren, Y.; Dai, L.; Guan, L.; Zhang, J.; Lin, X.; et al. Exome sequencing reveals mutation in GJA1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum. Mol. Genet. 2015, 24, 243–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heathcote, K.; Syrris, P.; Carter, N.D.; Patton, M.A. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J. Med. Genet. 2000, 37, 50–51. [Google Scholar] [CrossRef] [Green Version]
- Richard, G.; Smith, L.E.; Bailey, R.A.; Itin, P.; Hohl, D.; Epstein, E.H., Jr.; DiGiovanna, J.J.; Compton, J.G.; Bale, S.J. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat. Genet. 1998, 20, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Macari, F.; Landau, M.; Cousin, P.; Mevorah, B.; Brenner, S.; Panizzon, R.; Schorderet, D.F.; Hohl, D.; Huber, M. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am. J. Hum. Genet. 2000, 67, 1296–1301. [Google Scholar] [CrossRef]
- Lamartine, J.; Essenfelder, G.M.; Kibar, Z.; Lanneluc, I.; Callouet, E.; Laoudj, D.; Lemaitre, G.; Hand, C.; Haylick, S.J.; Zonana, J.; et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat. Genet. 2000, 26, 142–144. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Ramot, Y.; Molho-Pessach, V.; Meir, T.; Alper-Pinus, R.; Siam, I.; Tams, S.; Babay, S.; Zlotogorski, A. Mutation in KANK2, encoding a sequestering protein for steroid receptor coactivators, causes keratoderma and woolly hair. J. Med. Genet. 2014, 51, 388–394. [Google Scholar] [CrossRef]
- Kimonis, V.; DiGiovanna, J.J.; Yang, J.M.; Doyle, S.Z.; Bale, S.J.; Compton, J.G. A mutation in the V1 end domain of keratin 1 in non-epidermolytic palmar-plantarkeratoderma. J. Investig. Dermatol. 1994, 103, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Bowden, P.E.; Haley, J.L.; Kansky, A.; Rothnagel, J.A.; Jones, D.O.; Turner, R.J. Mutation of a type II keratin gene (K6a) in pachyonychia congenita. Nat. Genet. 1995, 10, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.J.D.; Jonkman, M.F.; van Goor, H.; Coleman, C.M.; Covello, S.P.; Uitto, J.; McLean, W.H.I. A mutation in human keratin K6b produces a phenocopy of the K17 disorder pachyonychia congenita type 2. Hum. Mol. Genet. 1998, 7, 1143–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.J.; Messenger, A.G.; Leachman, S.A.; O’Toole, E.A.; Lane, E.B.; McLean, W.H.I.; Smith, F.J.D. Keratin K6c mutations cause focal palmoplantar keratoderma. J. Investig. Derm. 2010, 130, 425–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, A.; Hennies, H.-C.; Langbein, L.; Digweed, M.; Mischke, D.; Drechsler, M.; Schrock, E.; Royer-Pokora, B.; Franke, W.W.; Sperling, K.; et al. Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma (EPPK). Nat. Genet. 1994, 6, 174–179. [Google Scholar] [CrossRef]
- Shamsher, M.K.; Navsaria, H.A.; Stevens, H.P.; Ratnavel, R.C.; Purkis, P.E.; McLean, W.H.; Cook, L.J.; Griffiths, W.A.D.; Geschmeissner, S.; Spurr, N.; et al. Novel mutations in keratin 16 gene underly focal nonepidermolytic palmoplantar keratoderma (NEPPK) in two families. Hum. Mol. Genet. 1995, 4, 1875–1881. [Google Scholar] [CrossRef]
- McLean, W.H.I.; Rugg, E.L.; Lunny, D.P.; Morley, S.M.; Lane, E.B.; Swensson, O.; Dopping-Hepenstal, P.J.C.; Griffiths, W.A.D.; Eady, R.A.J.; Higgins, C.; et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat. Genet. 1995, 9, 273–278. [Google Scholar] [CrossRef]
- Maestrini, E.; Monaco, A.P.; McGrath, J.A.; Ishida-Yamamoto, A.; Camisa, C.; Hovnanian, A.; Weeks, D.E.; Lathrop, M.; Uitto, J.; Christiano, A.M. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel’s syndrome. Nat. Genet. 1996, 13, 70–77. [Google Scholar] [CrossRef]
- Dahlqvist, J.; Klar, J.; Tiwari, N.; Schuster, J.; Torma, H.; Badhai, J.; Pujol, R.; van Steensel, M.A.M.; Brinkhuizen, T.; Gijezen, L.; et al. A single-nucleotide deletion in the POMP 5-prime UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am. J. Hum. Genet. 2010, 86, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Courcet, J.-B.; Elalaoui, S.C.; Duplomb, L.; Tajir, M.; Riviere, J.-B.; Thevenon, J.; Gigot, N.; Marle, N.; Aral, B.; Duffourd, Y.; et al. Autosomal-recessive SASH1 variants associated with a new genodermatosis with pigmentation defects, palmoplantar keratoderma and skin carcinoma. Eur. J. Hum. Genet. 2015, 23, 957–962. [Google Scholar]
- Kubo, A.; Shiohama, A.; Sasaki, T.; Nakabayashi, K.; Kawasaki, H.; Atsugi, T.; Sato, S.; Shimizu, A.; Mikami, S.; Tanizaki, H.; et al. Mutations in SERPINB7, encoding a member of the serine protease inhibitor superfamily, cause Nagashima-type palmoplantar keratosis. Am. J. Hum. Genet. 2013, 93, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Bouadjar, B.; Heilig, R.; Huber, M.; Lefevre, C.; Jobard, F.; Macari, F.; Bakija-Konsuo, A.; Ait-Belkacem, F.; Weissenbach, J.; et al. Mutations in the gene encoding SLURP-1 in mal de Meleda. Hum. Mol. Genet. 2001, 10, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natt, E.; Kida, K.; Odievre, M.; Di Rocco, M.; Scherer, G. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc. Nat. Acad. Sci. USA 1992, 89, 9297–9301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.; Rettler, I.; Bernasconi, K.; Frenk, E.; Lavrijsen, S.P.M.; Ponec, M.; Bon, A.; Lautenschlager, S.; Schorderet, D.F.; Hohl, D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science 1995, 267, 525–528. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, K.; Zhang, X.; Chen, Q.; Wu, J.; Li, H.; Zhou, Y.; Glusman, G.; Roach, J.; Etheridge, A.; et al. A gain-of-function mutation in TRPV3 causes focal palmoplantar keratoderma in a Chinese family. J. Investig. Derm. 2015, 135, 907–909. [Google Scholar] [CrossRef] [Green Version]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Drögemüller, M.; Jagannathan, V.; Becker, D.; Drögemüller, C.; Schelling, C.; Plassais, J.; Kaerle, C.; Dufaure de Citres, C.; Thomas, A.; Müller, E.J.; et al. A mutation in the FAM83G gene in dogs with hereditary footpad hyperkeratosis (HFH). PLoS Genet. 2014, 10, e1004370. [Google Scholar] [CrossRef]
- Balmer, P.; Fellay, A.K.; Sayar, B.S.; Hariton, W.V.J.; Wiener, D.J.; Galichet, A.; Müller, E.J.; Roosje, P.J. FAM83G/Fam83g genetic variants affect canine and murine hair formation. Exp. Dermatol. 2019, 28, 350–354. [Google Scholar] [CrossRef]
- Vogt, J.; Dingwell, K.S.; Herhaus, L.; Gourlay, R.; Macartney, T.; Campbell, D.; Smith, J.C.; Sapkota, G.P. Protein associated with SMAD1 (PAWS1/FAM83G) is a substrate for type I bone morphogenetic protein receptors and modulates bone morphogenetic protein signalling. Open Biol. 2014, 4, 130210. [Google Scholar] [CrossRef] [Green Version]
- Bozatzi, P.; Dingwell, K.S.; Wu, K.Z.; Cooper, F.; Cummins, T.D.; Hutchinson, L.D.; Vogt, J.; Wood, N.T.; Macartney, T.J.; Varghese, J.; et al. PAWS1 controls Wnt signalling through association with casein kinase 1α. EMBO Rep. 2018, 19, e44807. [Google Scholar] [CrossRef]
- Radden, L.A.; Child, K.M.; Adkins, E.B.; Spacek, D.V.; Feliciano, A.M.; King, T.R. The wooly mutation (wly) on mouse chromosome 11 is associated with a genetic defect in Fam83g. BMC Res. Notes 2013, 6, 189. [Google Scholar] [CrossRef] [Green Version]
- Plassais, J.; Guaguère, E.; Lagoutte, L.; Guillory, A.S.; de Citres, C.D.; Degorce-Rubiales, F.; Delverdier, M.; Vaysse, A.; Quignon, P.; Bleuart, C.; et al. A spontaneous KRT16 mutation in a dog breed: A model for human focal non-epidermolytic palmoplantar keratoderma (FNEPPK). J. Investig. Dermatol. 2015, 135, 1187–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Najor, N.A.; Green, K.J. Desmosomes: Regulators of cellular signaling and adhesion in epidermal health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, a015297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koulu, L.; Kusumi, A.; Steinberg, M.S.; Klaus-Kovtun, V.; Stanley, J.R. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J. Exp. Med. 1984, 160, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.M.; Rickman, L.; Whittock, N.V.; Eady, R.A.; Simrak, D.; Dopping-Hepenstal, P.J.; Stevens, H.P.; Armstrong, D.K.; Hennies, H.C.; Küster, W.; et al. Spectrum of dominant mutations in the desmosomal cadherin desmoglein 1, causing the skin disease striate palmoplantar keratoderma. Eur. J. Hum. Genet. 2001, 9, 197–203. [Google Scholar] [CrossRef]
- Samuelov, L.; Sarig, O.; Harmon, R.M.; Rapaport, D.; Ishida-Yamamoto, A.; Isakov, O.; Koetsier, J.L.; Gat, A.; Goldberg, I.; Bergman, R.; et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat. Genet. 2013, 45, 1244–1248. [Google Scholar] [CrossRef] [Green Version]
- Has, C.; Jakob, T.; He, Y.; Kiritsi, D.; Hausser, I.; Bruckner-Tuderman, L. Loss of desmoglein 1 associated with palmoplantar keratoderma, ermatitis and multiple allergies. Br. J. Dermatol. 2015, 172, 257–261. [Google Scholar] [CrossRef]
- Schlipf, N.A.; Vahlquist, A.; Teigen, N.; Virtanen, M.; Dragomir, A.; Fismen, S.; Barenboim, M.; Manke, T.; Rösler, B.; Zimmer, A.; et al. Whole-exome sequencing identifies novel autosomal recessive DSG1 mutations associated with mild SAM syndrome. Br. J. Dermatol. 2016, 174, 444–448. [Google Scholar] [CrossRef]
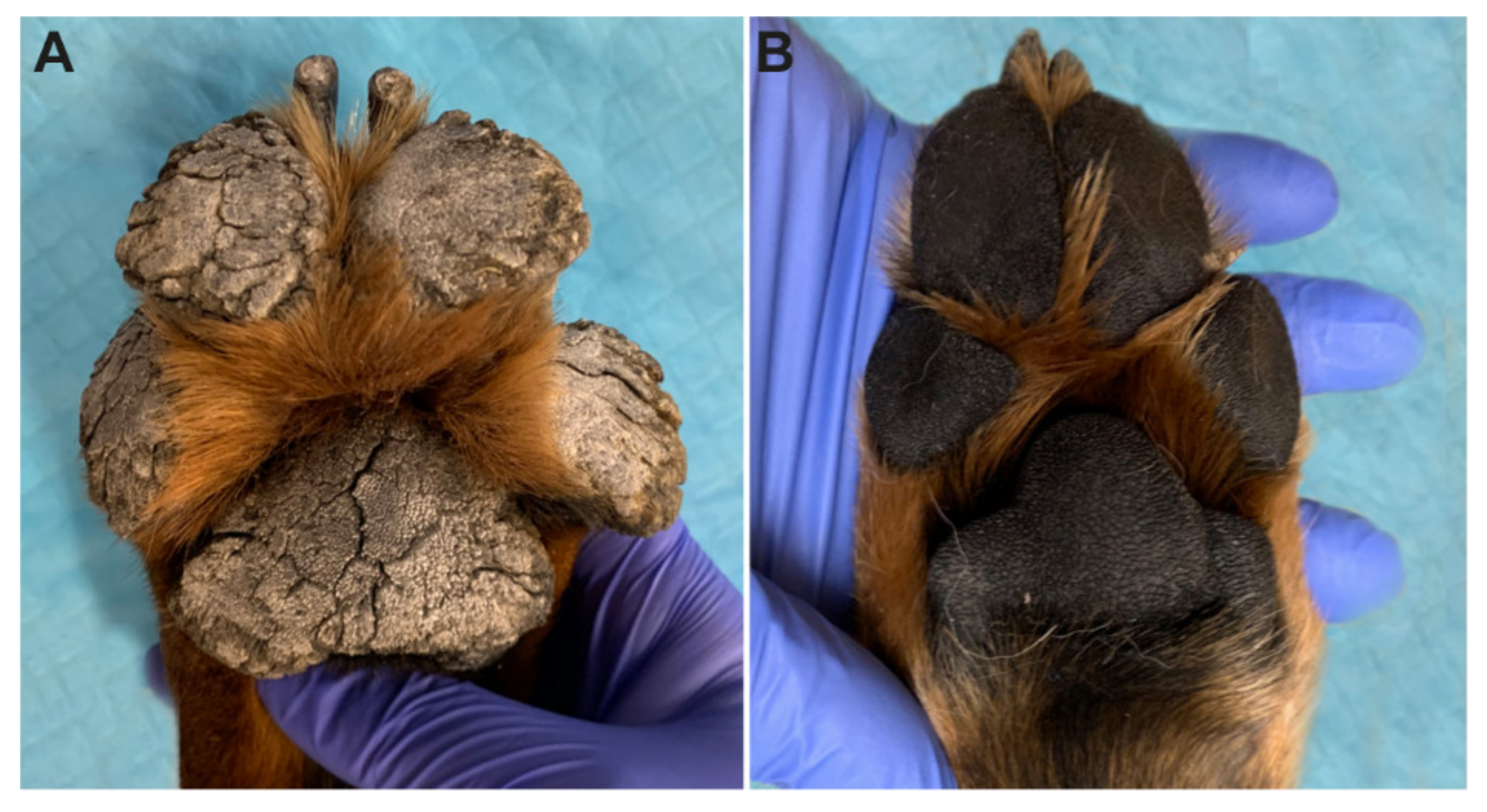
Figure 1.
Clinical phenotype at six months of age. (A) Marked expansion of the footpads by thick projections of dense keratin in the affected Rottweiler. The adjacent haired skin appears unaffected on this image. (B) Footpads of a normal six-month-old control Rottweiler.
Figure 1.
Clinical phenotype at six months of age. (A) Marked expansion of the footpads by thick projections of dense keratin in the affected Rottweiler. The adjacent haired skin appears unaffected on this image. (B) Footpads of a normal six-month-old control Rottweiler.

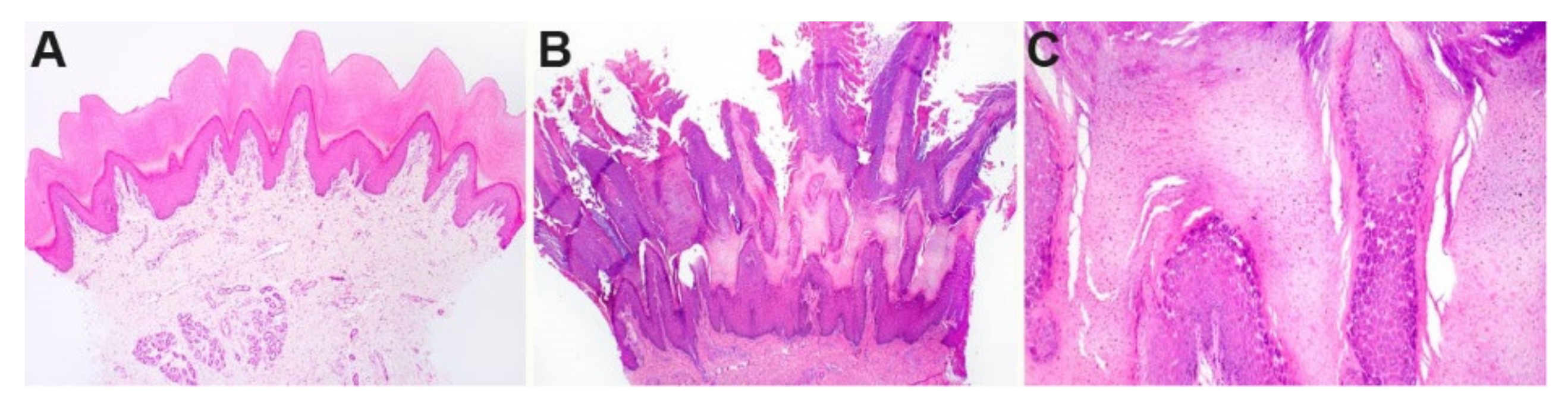
Figure 2.
Histopathological phenotype of footpad skin. (A) Footpad skin of a normal six-month-old control dog. (B) In a biopsy taken from the affected dog at six months of age, a dense proliferation of the stratum corneum (outermost anucleated layer of the skin) markedly expands the epidermis. The stratum corneum is arranged in papillated villous projections of dense keratin. (C) Higher magnification of the stratum granulosum and stratum corneum of the affected dog shows an expansion of the granular cell layer.
Figure 2.
Histopathological phenotype of footpad skin. (A) Footpad skin of a normal six-month-old control dog. (B) In a biopsy taken from the affected dog at six months of age, a dense proliferation of the stratum corneum (outermost anucleated layer of the skin) markedly expands the epidermis. The stratum corneum is arranged in papillated villous projections of dense keratin. (C) Higher magnification of the stratum granulosum and stratum corneum of the affected dog shows an expansion of the granular cell layer.

Figure 3.
Details of the DSG1:c.2541_2545delGGGCT variant. Representative electropherograms of a control and the affected dog are shown. The amino acid translations of the wild-type and mutant alleles are indicated.
Figure 3.
Details of the DSG1:c.2541_2545delGGGCT variant. Representative electropherograms of a control and the affected dog are shown. The amino acid translations of the wild-type and mutant alleles are indicated.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of genetic causes of human palmoplantar keratodermas (PPK).
| Gene | Phenotype | Inheritance a | Ref. |
|---|---|---|---|
| AAGAB | Palmoplantar keratoderma, punctate type IA; PPKP1A | AD | [3,4] |
| AQP5 | Palmoplantar keratoderma, Bothnian type | AD | [5] |
| CARD5 | Pityriasis rubra pilaris | AD | [6] |
| COL14A1 | Palmoplantar keratoderma, punctate type IB; PPKP1B | AD | [7] |
| CTSC | Papillon-Lefevre syndrome | AR | [8] |
| DSG1 | Palmoplantar keratoderma I, striate, focal, or diffuse; PPKS1 | AD | [9] |
| DSP | Palmoplantar keratoderma II, striate, focal, or diffuse; PPKS2 | AD | [10] |
| ENPP1 | Cole disease | AD | [11] |
| FAM83G | Palmoplantar keratoderma and exuberant scalp hair. | AR | [12] |
| GJA1 | Palmoplantar keratoderma with congenital alopecia | AD | [13] |
| GJB2 | Keratoderma, palmoplantar, with deafness | AD | [14] |
| GJB3 | Erythrokeratodermia variabilis et progressiva 1 | AD or AR | [15] |
| GJB4 | Erythrokeratodermia variabilis et progressiva 2 | AD | [16] |
| GJB6 | Ectodermal dysplasia 2, Clouston type | AD | [17] |
| JUP | Naxos disease | AR | [18] |
| KANK2 | Palmoplantar keratoderma and woolly hair | AR | [19] |
| KRT1 | Palmoplantar keratoderma, epidermolytic or nonepidermolytic | AD | [20] |
| KRT6A | Pachyonychia congenita 3 | AD | [21] |
| KRT6B | Pachyonychia congenita 4 | AD | [22] |
| KRT6C | Palmoplantar keratoderma, nonepidermolytic, focal or diffuse | AD | [23] |
| KRT9 | Palmoplantar keratoderma, epidermolytic | AD | [24] |
| KRT16 | Palmoplantar keratoderma, nonepidermolytic, focal 1, FNEPPK1 | AD | [25,26] |
| KRT17 | Pachyonychia congenita 2 | AD | [26] |
| LOR | Vohwinkel syndrome with ichthyosis | AD | [27] |
| POMP | Keratosis linearis with ichthyosis congenita and sclerosing keratoderma | AR | [28] |
| SASH1 | Cancer, alopecia, pigment dyscrasia, onychodystrophy, and keratoderma | AR | [29] |
| SERPINB7 | Palmoplantar keratoderma, Nagashima type; PPKN | AR | [30] |
| SLURP1 | Meleda disease | AR | [31] |
| TAT | Tyrosinemia, type II | AR | [32] |
| TGM1 | Ichthyosis, congenital, autosomal recessive 1 | AR | [33] |
| TRPV3 | Palmoplantar keratoderma, nonepidermolytic, focal 2; FNEPPK2 | AD | [34] |
| WNT10A | Schöpf–Schulz–Passarge syndrome | AR | [35] |
a AD: autosomal dominant; AR: autosomal recessive.
Table 2.
Results of variant filtering in the affected Rottweiler dog against 655 control genomes.
| Filtering Step | Homozygous Variants | Heterozygous Variants |
|---|---|---|
| All variants in the affected Rottweiler | 3,310,269 | 2,516,875 |
| Private variants | 842 | 3290 |
| Protein-changing private variants | 4 | 25 |
| Private variants in known candidate genes | 1 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Backel, K.A.; Kiener, S.; Jagannathan, V.; Casal, M.L.; Leeb, T.; Mauldin, E.A. A DSG1 Frameshift Variant in a Rottweiler Dog with Footpad Hyperkeratosis. Genes 2020, 11, 469. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040469
AMA Style
Backel KA, Kiener S, Jagannathan V, Casal ML, Leeb T, Mauldin EA. A DSG1 Frameshift Variant in a Rottweiler Dog with Footpad Hyperkeratosis. Genes. 2020; 11(4):469. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040469
Chicago/Turabian StyleBackel, Katherine A., Sarah Kiener, Vidhya Jagannathan, Margret L. Casal, Tosso Leeb, and Elizabeth A. Mauldin. 2020. "A DSG1 Frameshift Variant in a Rottweiler Dog with Footpad Hyperkeratosis" Genes 11, no. 4: 469. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040469
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.