DNA Hypermethylation and Unstable Repeat Diseases: A Paradigm of Transcriptional Silencing to Decipher the Basis of Pathogenic Mechanisms



Abstract
:
1. Introduction
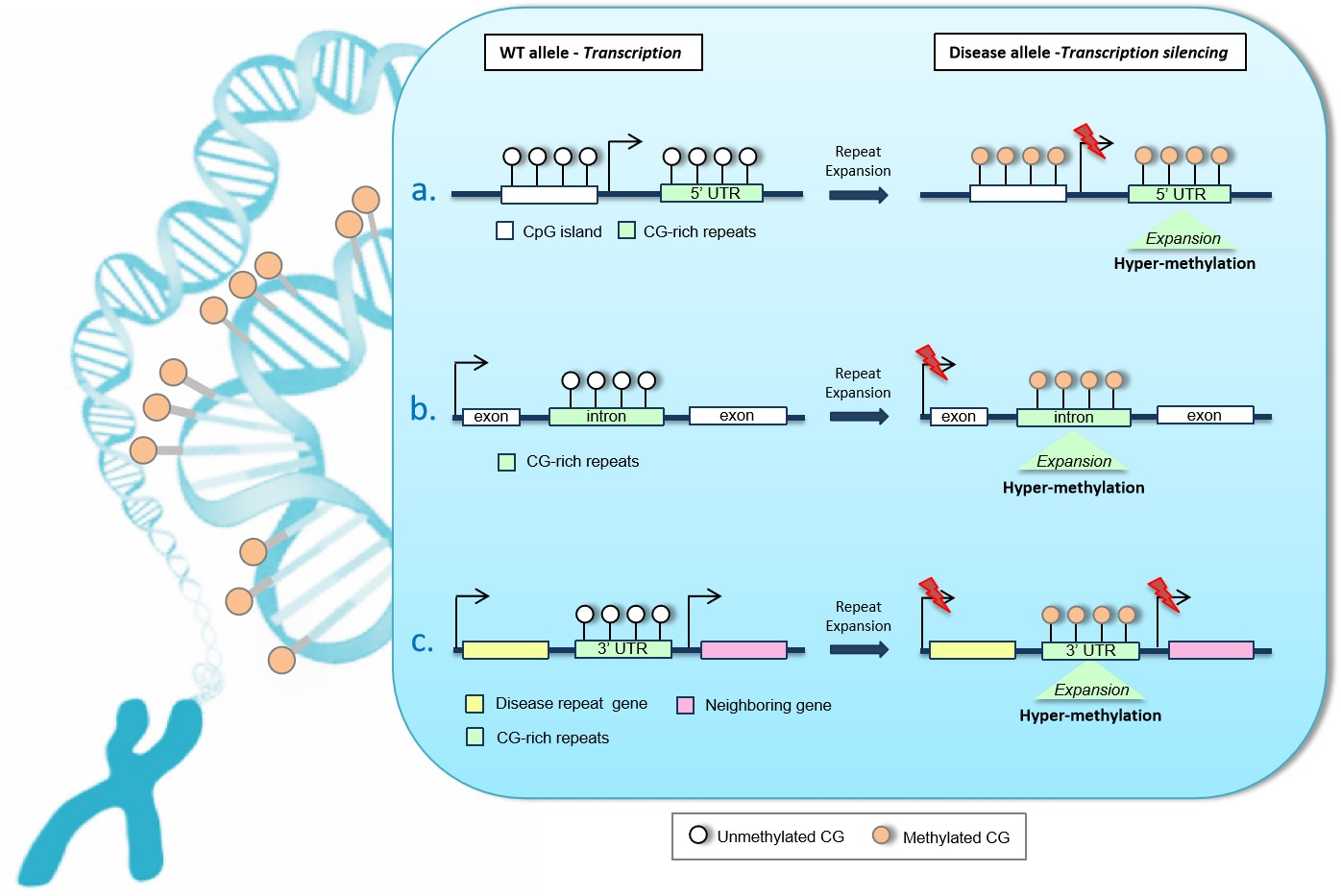
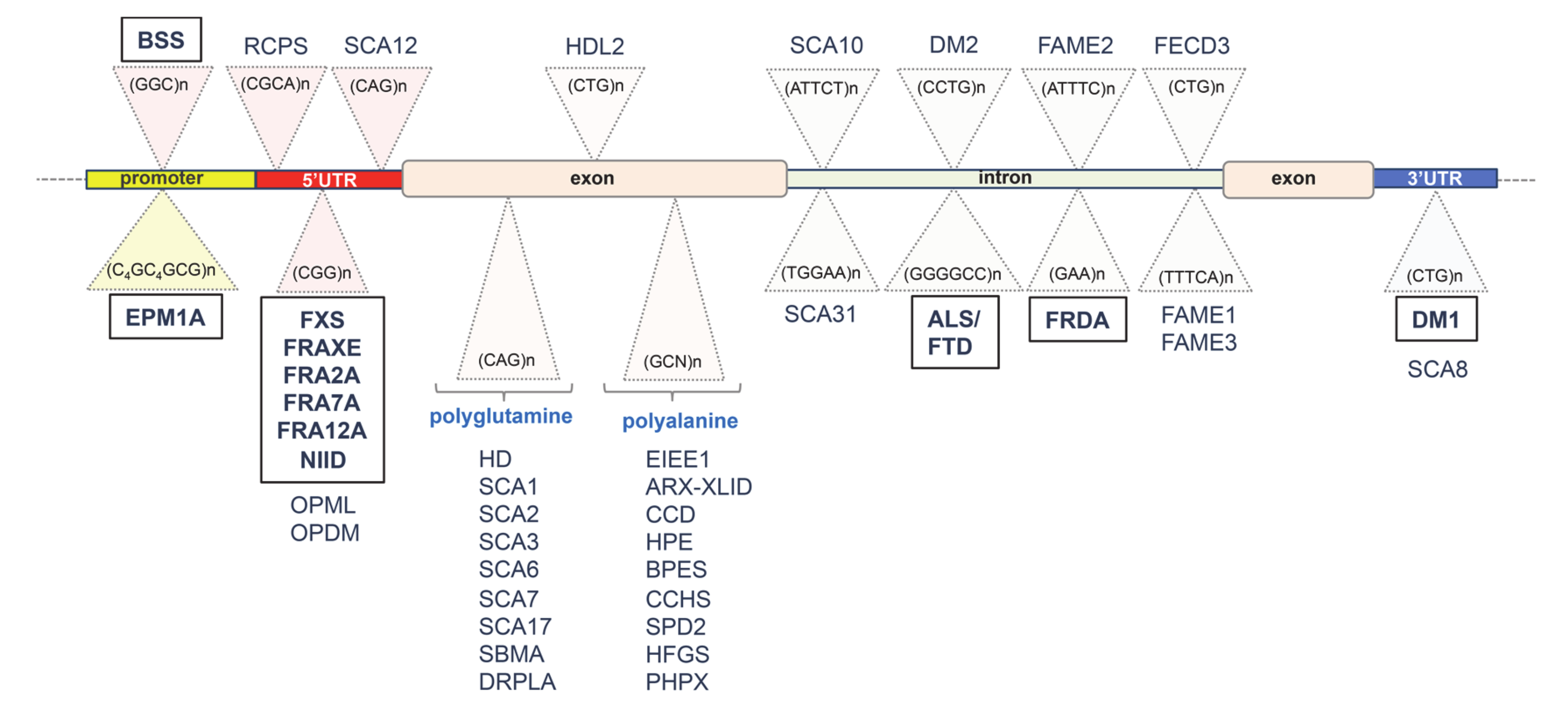
2. Unstable GC-Rich Repeats in 5′ Regulatory Regions
2.1. GGC Repeat Expansion with Hypermethylation of XYLT1 Exon 1 in Baratela–Scott Syndrome (BSS)
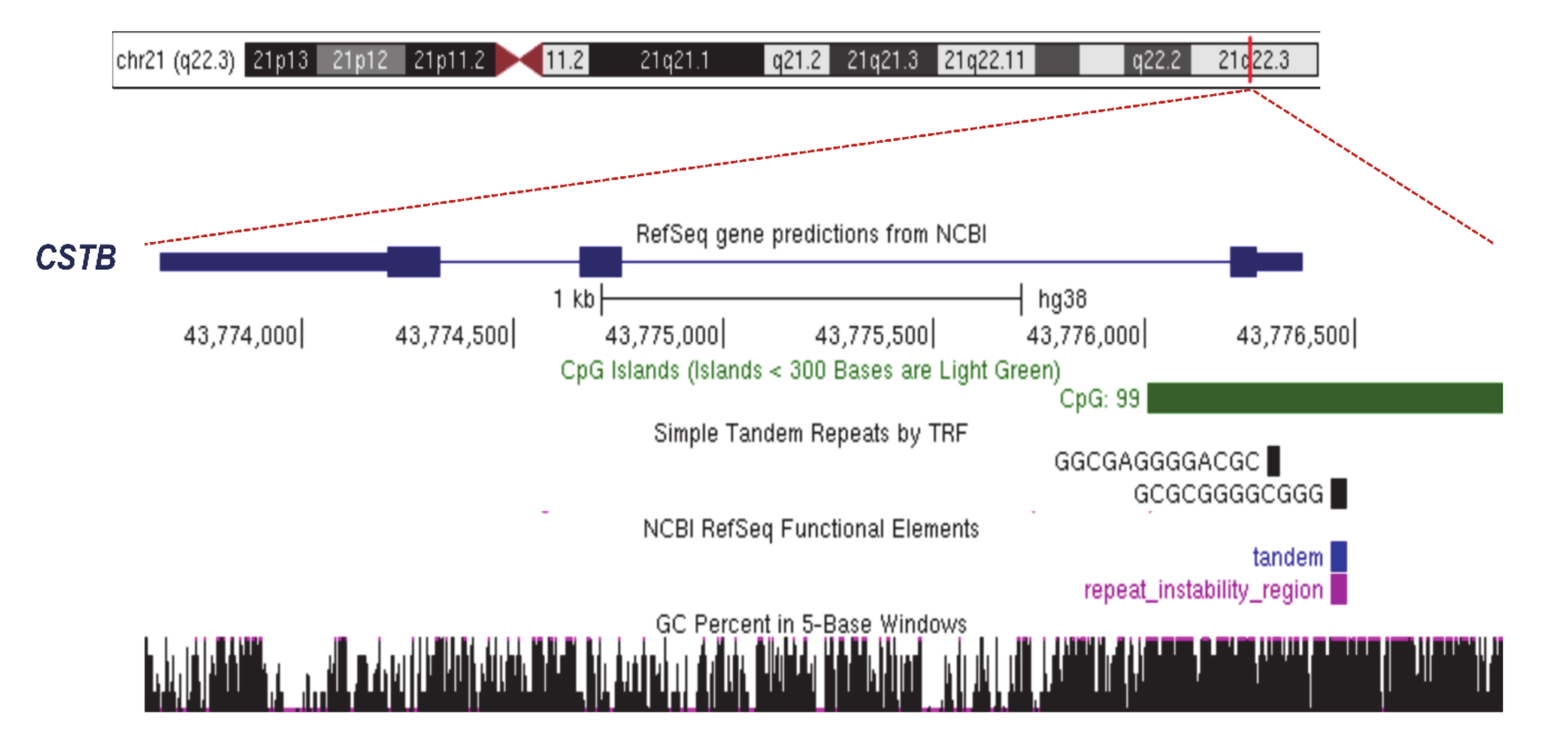
2.2. Unstable Dodecamer Repeat in Progressive Myoclonus Epilepsy of Unverricht–Lundborg Type 1A (EPM1A)
2.3. Hypermethylated CGG Repeats in Fragile X Syndrome (FXS)
2.4. Beyond the FMR1 Locus: Hypermethylated GC-Rich Repeats in Rare Folate-Sensitive Fragile Sites (FSFS)
2.5. CGG Repeat Instability in Familial Neuronal Intranuclear Inclusion Disease (NIID)
3. Unstable Hypermethylated Repeats in Intronic Regions
3.1. GGGGCC Expanded Repeat in Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD)
3.2. Intronic GAA Repeat Expansion in Friedreich Ataxia (FRDA)
4. Unstable GC-Rich Repeats in 3′ Untranslated Region (3′UTR)
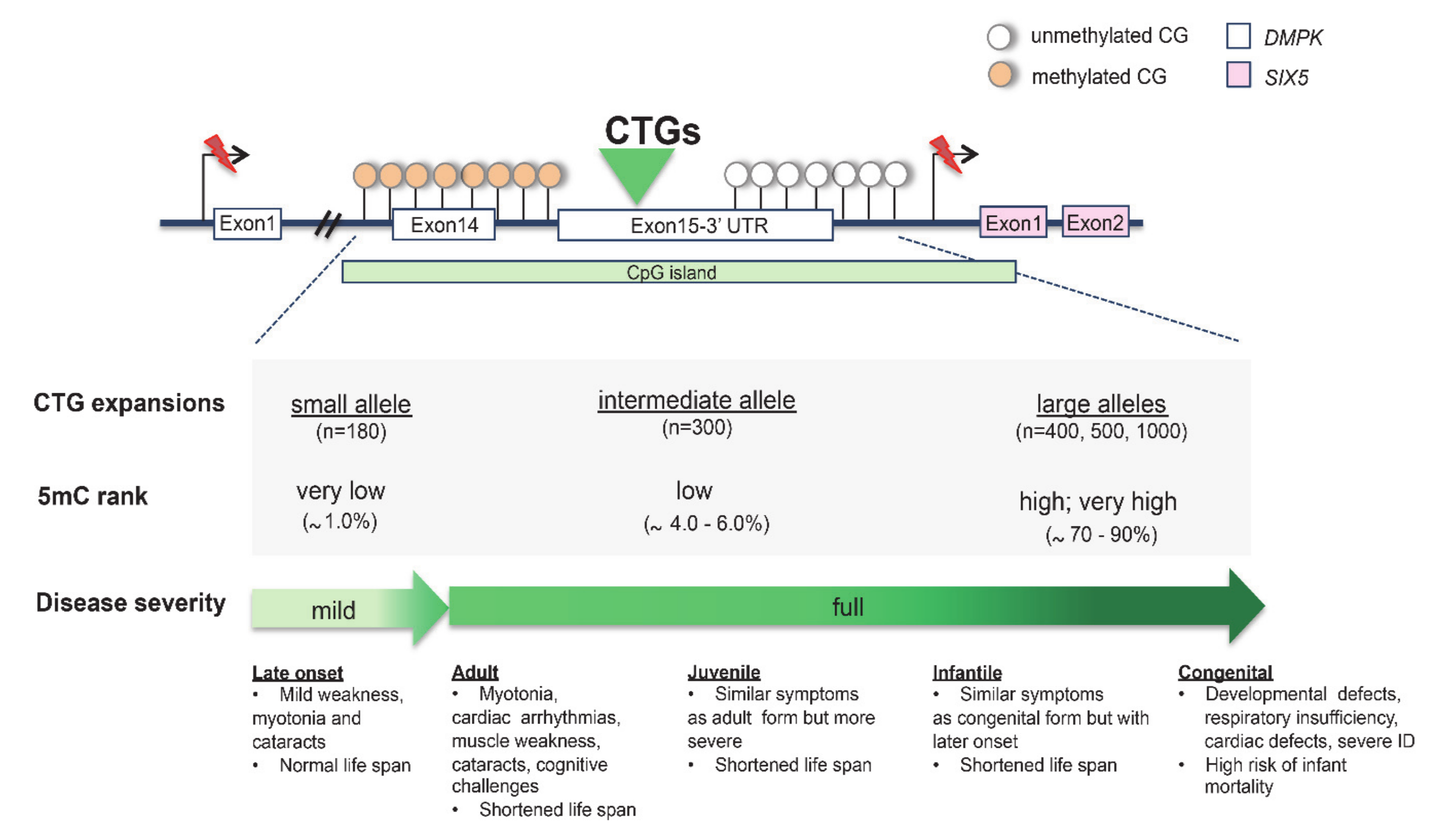
CTG Triplet Expansion in Myotonic Dystrophy Type 1 (DM1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Repeat | Gene | Locus/Disease | Inheritance | Hyper-Methylation | Changes in 5hmC | Reference |
|---|---|---|---|---|---|---|---|
| Promoter | |||||||
| (GGC)n | XYLT1 | BSS | AR | Yes | (?) | [22] | |
| (C4GC4GCG)n | CSTB | EPM1A | AR | (?) | (?) | [28,31] | |
| 5′ UTR | (CGG)n | FMR1 | FXS | XL | Yes | Yes | [16,33,36,37,43] |
| AFF2 | FRAXE | XL | Yes | (?) | [46] | ||
| AFF3 | FRA2A | AD | Yes | (?) | [44] | ||
| ZNF713 | FRA7A | AD | Yes | (?) | [45] | ||
| DIP2B | FRA12A | AD | Yes | (?) | [50] | ||
| NOTCH2NLC | NIID | AD | Yes | (?) | [51] | ||
| intron | (GGGGCC)n | C9orf72 | ALS/FTD | AD | Yes | Yes | [10,60] |
| (GAA)n | FXN | FRDA | AR | Yes | Yes | [11,17,68] | |
| 3′ UTR | (CTG)n | DMPK | DM1 | AD | Yes | (?) | [74] |
5. Diagnostic Tools
6. Therapeutic Approaches
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, R.; Nagpal, G.; Kumar, V.; Usmani, S.S.; Agrawal, P.; Raghava, G.P.S. HumCFS: A database of fragile sites in human chromosomes. BMC Genom. 2019, 19, 985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poeta, L.; Fusco, F.; Drongitis, D.; Shoubridge, C.; Manganelli, G.; Filosa, S.; Paciolla, M.; Courtney, M.; Collombat, P.; Lioi, M.B.; et al. A Regulatory Path Associated with X-Linked Intellectual Disability and Epilepsy Links KDM5C to the Polyalanine Expansions in ARX. Am. J. Hum. Genet. 2013, 92, 114–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanni, S.; Pearson, C.E. Molecular genetics of congenital myotonic dystrophy. Neurobiol. Dis. 2019, 132, 104533. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef]
- Nelson, D.L.; Orr, H.T.; Warren, S. The Unstable Repeats—Three Evolving Faces of Neurological Disease. Neuron 2013, 77, 825–843. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium; 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Shoubridge, C.; Fullston, T.; Gécz, J. ARX spectrum disorders: Making inroads into the molecular pathology. Hum. Mutat. 2010, 31, 889–900. [Google Scholar] [CrossRef]
- Laperuta, C.; Spizzichino, L.; D’Adamo, P.; Monfregola, J.; Maiorino, A.; D’Eustacchio, A.; Ventruto, V.; Neri, G.; D’Urso, M.; Chiurazzi, P.; et al. MRX87 family with Aristaless X dup24bp mutation and implication for polyAlanine expansions. BMC Med. Genet. 2007, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Cattaneo, E. Huntington’s disease. Handb. Exp. Pharmacol. 2014, 220, 357–409. [Google Scholar]
- Pattamatta, A.; Cleary, J.D.; Ranum, L.P. All in the Family: Repeats and ALS/FTD. Trends Neurosci. 2018, 41, 247–250. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Bidichandani, S.I. Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 2019, 132, 104606. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.N. Genetics and Epigenetics of the Multifunctional Protein CTCF. Current. Top. Dev. Biol. 2007, 80, 337–360. [Google Scholar] [CrossRef]
- Groh, M.; Lufino, M.M.P.; Wade-Martins, R.; Gromak, N. R-loops Associated with Triplet Repeat Expansions Promote Gene Silencing in Friedreich Ataxia and Fragile X Syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nageshwaran, S.; Festenstein, R. Epigenetics and Triplet-Repeat Neurological Diseases. Front. Neurol. 2015, 6, 1010. [Google Scholar] [CrossRef] [Green Version]
- Madrid, A.; Papale, L.A.; Alisch, R.S. New hope: The emerging role of 5-hydroxymethylcytosine in mental health and disease. Epigenomics 2016, 8, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Brasa, S.; Mueller, A.; Jacquemont, S.; Hahne, F.; Rozenberg, I.; Peters, T.; He, Y.; McCormack, C.; Gasparini, F.; Chibout, S.-D.; et al. Reciprocal changes in DNA methylation and hydroxymethylation and a broad repressive epigenetic switch characterize FMR1 transcriptional silencing in fragile X syndrome. Clin. Epigenetics 2016, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Sandi, C.; Pinto, R.M.; Pook, M.A. Friedreich Ataxia Patient Tissues Exhibit Increased 5-Hydroxymethylcytosine Modification and Decreased CTCF Binding at the FXN Locus. PLoS ONE 2013, 8, e74956. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Cheng, Y.; Bernstein, A.; Chen, D.; Jin, P. 5-Hydroxymethylcytosine: A new player in brain disorders? Exp. Neurol. 2015, 268, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Khare, T.; Pai, S.; Koncevičius, K.; Pal, I.; Kriukienė, E.; LiutkeviČiŪtĖ, Z.; Irimia, M.; Jia, P.; Ptak, C.; Xia, M.; et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Boil. 2012, 19, 1037–1043. [Google Scholar] [CrossRef] [Green Version]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000, 113, 193–205. [Google Scholar] [PubMed]
- Lacroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokhman, D.; Nissim-Rafinia, M.; Agranat-Tamir, L.; Housman, G.; García-Pérez, R.; Lizano, E.; Cheronet, O.; Mallick, S.; Nieves-Colón, M.A.; Li, H.; et al. Differential DNA methylation of vocal and facial anatomy genes in modern humans. Nat. Commun. 2020, 11, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, N.; Girard, J.-M.; Turnbull, J.; A Minassian, B. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia 2009, 50, 29–36. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Lehesjoki, A.-E.; Stone, N.E.; Willour, V.L.; Virtaneva, K.; Miao, J.; D’Amato, E.; Ramirez, L.; Faham, M.; Koskiniemi, M.; et al. Mutations in the Gene Encoding Cystatin B in Progressive Myoclonus Epilepsy (EPM1). Science 1996, 271, 1731–1734. [Google Scholar] [CrossRef]
- Penna, E.; Cerciello, A.; Chambery, A.; Russo, R.; Cernilogar, F.M.; Pedone, E.M.; Perrone-Capano, C.; Cappello, S.; Di Giaimo, R.; Crispino, M. Cystatin B Involvement in Synapse Physiology of Rodent Brains and Human Cerebral Organoids. Front. Mol. Neurosci. 2019, 12, 195. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, T.; Kuronen, M.; Alakurtti, K.; Tegelberg, S.; Hakala, P.; Aalto, A.; Huopaniemi, L.; Aula, N.; Michellucci, R.; Eriksson, K.; et al. Cystatin B: Mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients. Eur. J. Hum. Genet. 2006, 15, 185–193. [Google Scholar] [CrossRef]
- Alakurtti, K.; Virtaneva, K.; Joensuu, T.; Palvimo, J.J.; Lehesjoki, A.-E. Characterization of the cystatin B gene promoter harboring the dodecamer repeat expanded in progressive myoclonus epilepsy, EPM1. Genetics 2000, 242, 65–73. [Google Scholar] [CrossRef]
- O’Brien, A.; Marshall, C.R.; Blaser, S.; Ray, P.N.; Yoon, G. Severe neurodegeneration, progressive cerebral volume loss and diffuse hypomyelination associated with a homozygous frameshift mutation in CSTB. Eur. J. Hum. Genet. 2017, 25, 775–778. [Google Scholar] [CrossRef]
- Weinhaeusel, A.; Morris, M.A.; Antonarakis, S.E.; Haas, O.A. DNA deamination enables direct PCR amplification of the cystatin B (CSTB) gene-associated dodecamer repeat expansion in myoclonus epilepsy type Unverricht-Lundborg. Hum. Mutat. 2003, 22, 404–408. [Google Scholar] [CrossRef]
- Horiuchi, H.; Osawa, M.; Furutani, R.; Morita, M.; Tian, W.; Awatsu, Y.; Shimazaki, H.; Umetsu, K. Polymerase Chain Reaction-Based Analysis Using Deaminated DNA of Dodecamer Expansions in CSTB, Associated with Unverricht-Lundborg Myoclonus Epilepsy. Genet. Test. 2005, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Voutsinos, V.; Munk, S.H.N.; Oestergaard, V.H. Common Chromosomal Fragile Sites—Conserved Failure Stories. Genes 2018, 9, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, F.R.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.-L.; et al. Fragile X syndrome. Nat. Rev. Dis. Prim. 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J.; Hagerman, R. Fragile X-associated Tremor/Ataxia Syndrome (FXTAS). Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 25–30. [Google Scholar] [CrossRef]
- Miano, M.G.; Laperuta, C.; Chiurazzi, P.; D’Urso, M.; Ursini, M.V. Ovarian dysfunction and FMR1 alleles in a large Italian family with POF and FRAXA disorders: Case report. BMC Med. Genet. 2007, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Naumann, A.; Hochstein, N.; Weber, S.; Fanning, E.; Doerfler, W. A Distinct DNA-Methylation Boundary in the 5′- Upstream Sequence of the FMR1 Promoter Binds Nuclear Proteins and Is Lost in Fragile X Syndrome. Am. J. Hum. Genet. 2009, 85, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabolacci, E.; Moscato, U.; Zalfa, F.; Bagni, C.; Chiurazzi, P.; Neri, G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008, 16, 1487–1498. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.M.; Wright, S.E.; Kearse, M.G.; Haenfler, J.M.; Flores, B.N.; Liu, Y.; Ifrim, M.F.; Glineburg, M.R.; Krans, A.; Jafar-Nejad, P.; et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat. Neurosci. 2020, 23, 386–397. [Google Scholar] [CrossRef]
- Baker, E.K.; Arpone, M.; Vera, S.A.; Bretherton, L.; Kraan, C.M.; Bui, M.; Slater, H.; Ling, L.; Francis, D.; Hunter, M.F.; et al. Incomplete silencing of full mutation alleles in males with fragile X syndrome is associated with autistic features. Mol. Autism 2019, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Kuznitsov-Yanovsky, L.; Mayshar, Y.; Ben-Yosef, D. Modeling FXS: Human Pluripotent Stem Cells and In Vitro Neural Differentiation; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; Volume 1942, pp. 89–100. [Google Scholar]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.-Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-Bound Trinucleotide Repeat mRNA Drives Epigenetic Silencing in Fragile X Syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [Green Version]
- Lanni, S.; Goracci, M.; Borrelli, L.; Mancano, G.; Chiurazzi, P.; Moscato, U.; Ferrè, F.; Helmer-Citterich, M.; Tabolacci, E.; Neri, G. Role of CTCF Protein in Regulating FMR1 Locus Transcription. PLoS Genet. 2013, 9, e1003601. [Google Scholar] [CrossRef] [Green Version]
- Esanov, R.; Andrade, N.S.; Bennison, S.; Wahlestedt, C.; Zeier, Z. The FMR1 promoter is selectively hydroxymethylated in primary neurons of fragile X syndrome patients. Hum. Mol. Genet. 2016, 25, 4870–4880. [Google Scholar] [CrossRef] [PubMed]
- Metsu, S.; Rooms, L.; Rainger, J.; Taylor, M.S.; Bengani, H.; Wilson, D.I.; Chilamakuri, C.S.R.; Morrison, H.; Vandeweyer, G.; Reyniers, E.; et al. FRA2A Is a CGG Repeat Expansion Associated with Silencing of AFF3. PLoS Genet. 2014, 10, e1004242. [Google Scholar] [CrossRef] [PubMed]
- Metsu, S.; Rainger, J.K.; Debacker, K.; Bernhard, B.; Rooms, L.; A Grafodatskaya, D.; Weksberg, R.; Fombonne, E.; Taylor, M.S.; Scherer, S.W.; et al. A CGG-Repeat Expansion Mutation inZNF713Causes FRA7A: Association with Autistic Spectrum Disorder in two Families. Hum. Mutat. 2014, 35, 1295–1300. [Google Scholar] [CrossRef]
- Gécz, J.; Gedeon, Á.K.; Sutherland, G.R.; Mulley, J.C. Identification of the gene FMR2, associated with FRAXE mental retardation. Nat. Genet. 1996, 13, 105–108. [Google Scholar] [CrossRef]
- Knight, S.; Flannery, A.; Hirst, M.; Campbell, L.; Christodoulou, Z.; Phelps, S.; Pointon, J.; Middleton-Price, H.; Barnicoat, A.; Pembrey, M.; et al. Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 1993, 74, 127–134. [Google Scholar] [CrossRef]
- Gu, Y.; McIlwain, K.L.; Weeber, E.J.; Yamagata, T.; Xu, B.; Antalffy, B.A.; Reyes, C.; Yuva-Paylor, L.; Armstrong, D.; Zoghbi, H.Y.; et al. Impaired Conditioned Fear and Enhanced Long-Term Potentiation inFmr2 Knock-Out Mice. J. Neurosci. 2002, 22, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Yuva-Aydemir, Y.; Almeida, S.; Krishnan, G.; Gendron, T.F.; Gao, F.B. Transcription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD. Nat. Commun. 2019, 10, 5466. [Google Scholar] [CrossRef]
- Winnepenninckx, B.; Debacker, K.; Ramsay, J.; Smeets, M.; Smits, A.; Fitzpatrick, D.R.; Kooy, R.F. CGG-Repeat Expansion in the DIP2B Gene Is Associated with the Fragile Site FRA12A on Chromosome 12q13.1. Am. J. Hum. Genet. 2006, 80, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [CrossRef]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.-L.; Huang, W.; Zeng, S.; Jiao, B.; Liu, Z.; Chen, Z.; Li, Y.; Wang, Y.; Min, H.-X.; et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 2019, 105, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.-Y.; Xu, Q.; Tian, Y.; Hu, Z.-M.; Qin, L.-X.; Yang, J.-X.; Huang, W.; Xue, J.; Li, J.-C.; Zeng, S.; et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain 2020, 143, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Belzil, V.V.; Zhang, Y.-J.; Petrucelli, L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014, 127, 359–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dols-Icardo, O.; García-Redondo, A.; Rojas-García, R.; Sanchez-Valle, R.; Noguera, A.; Gómez-Tortosa, E.; Pastor, P.; Hernández, I.; Esteban-Pérez, J.; Suárez-Calvet, M.; et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2013, 23, 749–754. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; MacKenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J.; et al. Hypermethylation of the CpG Island Near the G4C2 Repeat in ALS with a C9orf72 Expansion. Am. J. Hum. Genet. 2013, 92, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Belzil, V.V.; Katzman, R.B.; Petrucelli, L. ALS and FTD: An epigenetic perspective. Acta Neuropathol. 2016, 132, 487–502. [Google Scholar] [CrossRef]
- Esanov, R.; Belle, K.C.; Van Blitterswijk, M.; Belzil, V.V.; Rademakers, R.; Dickson, D.W.; Petrucelli, L.; Boylan, K.B.; Dykxhoorn, D.M.; Wuu, J.; et al. C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp. Neurol. 2016, 277, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Bauer, P.O. Methylation of C9orf72 expansion reduces RNA foci formation and dipeptide-repeat proteins expression in cells. Neurosci. Lett. 2016, 612, 204–209. [Google Scholar] [CrossRef]
- Liu, J.; Hu, J.; Ludlow, A.T.; Pham, J.T.; Shay, J.W.; Rothstein, J.D.; Corey, D.R. c9orf72 disease-related foci are each composed of one mutant expanded repeat RNA. Cell. Chem. Biol. 2017, 24, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans-Galea, M.V.; Carrodus, N.; Rowley, S.; Corben, L.A.; Tai, G.; Saffery, R.; Galati, J.C.; Wong, N.; Craig, J.M.; Lynch, D.R.; et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012, 71, 487–497. [Google Scholar] [CrossRef]
- Bradley, J.; Blake, J.; Chamberlain, S.; Thomas, P.; Cooper, J.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G.A. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaldo, I.; Pinelli, M.; Monticelli, A.; Acquaviva, F.; Giacchetti, M.; Filla, A.; Sacchetti, S.; Keller, S.; E Avvedimento, V.; Chiariotti, L.; et al. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med Genet. 2008, 45, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahdawi, S.; Pinto, R.M.; Ismail, O.; Varshney, D.; Lymperi, S.; Sandi, C.; Trabzuni, D.; Pook, M.A. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 2007, 17, 735–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, N.; Wang, L.; Lu, X.; Senut, M.-C.; Brenner, C.A.; Ruden, D.M. Hypothesis: Environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011, 6, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Tomé, S.; Gourdon, G. DM1 Phenotype Variability and Triplet Repeat Instability: Challenges in the Development of New Therapies. Int. J. Mol. Sci. 2020, 21, 457. [Google Scholar] [CrossRef] [Green Version]
- Barbé, L.; Lanni, S.; López-Castel, A.; Franck, S.; Spits, C.; Keymolen, K.; Seneca, S.; Tomé, S.; Miron, I.; Letourneau, J.; et al. CpG Methylation, a Parent-of-Origin Effect for Maternal-Biased Transmission of Congenital Myotonic Dystrophy. Am. J. Hum. Genet. 2017, 100, 488–505. [Google Scholar] [CrossRef] [Green Version]
- Yanovsky-Dagan, S.; Avitzour, M.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Mitrani-Rosenbaum, S.; Levy-Lahad, E.; Birnbaum, R.Y.; Gepstein, L.; et al. Uncovering the Role of Hypermethylation by CTG Expansion in Myotonic Dystrophy Type 1 Using Mutant Human Embryonic Stem Cells. Stem Cell Rep. 2015, 5, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Klesert, T.R.; Cho, D.H.; Clark, J.I.; Maylie, J.; Adelman, J.; Snider, L.; Yuen, E.C.; Soriano, P.; Tapscott, S.J. Mice deficient in Six5 develop cataracts: Implications for myotonic dystrophy. Nat. Genet. 2000, 25, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Castel, A.L.; Nakamori, M.; Tomé, S.; Chitayat, D.; Gourdon, G.; Thornton, C.A.; E Pearson, C. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 2010, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Légaré, C.; Overend, G.; Guay, S.-P.; Monckton, D.G.; Mathieu, J.; Gagnon, C.; Bouchard, L. DMPK gene DNA methylation levels are associated with muscular and respiratory profiles in DM1. Neurol. Genet. 2019, 5, e338. [Google Scholar] [CrossRef] [Green Version]
- Filippova, G.N.; Thienes, C.; Penn, B.H.; Cho, D.H.; Hu, Y.J.; Moore, J.M.; Klesert, T.R.; Lobanenkov, V.V.; Tapscott, S.J. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 2001, 28, 335–343. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, P.J.; Eid, J.; Peluso, P.; Rank, D.; Yin, J.; Hickey, L.; Tassone, F.; Loomis, E. Sequencing the Unsequenceable: Expanded CGG Repeats in the Human FMR1 Gene. Biophys. J. 2013, 104, 377a. [Google Scholar] [CrossRef] [Green Version]
- Zablotskaya, A.; Van Esch, H.; Verstrepen, K.J.; Froyen, G.; Vermeesch, J.R. Mapping the landscape of tandem repeat variability by targeted long read single molecule sequencing in familial X-linked intellectual disability. BMC Med Genom. 2018, 11, 123. [Google Scholar] [CrossRef] [Green Version]
- Cumming, S.; Hamilton, M.J.; Robb, Y.; Gregory, H.; McWilliam, C.; Cooper, A.; Adam, B.; McGhie, J.; Hamilton, G.; Herzyk, P.; et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur. J. Hum. Genet. 2018, 26, 1635–1647. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, M.-Y.; Wang, X.-J.; Hu, Z.-M.; Li, J.-C.; Li, N.; Wang, J.-L.; Liang, F.; Yang, Q.; Liu, Q.; et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J. Med Genet. 2018, 56, 265–270. [Google Scholar] [CrossRef]
- Dashnow, H.; Lek, M.; Phipson, B.; Halman, A.; Sadedin, S.; Lonsdale, A.; Davis, M.; Lamont, P.; Clayton, J.; Laing, N.G.; et al. STRetch: Detecting and discovering pathogenic short tandem repeat expansions. Genome Biol. 2018, 19, 121. [Google Scholar] [CrossRef] [Green Version]
- Genovese, L.M.; Geraci, F.; Corrado, L.; Mangano, E.; D’Aurizio, R.; Bordoni, R.; Severgnini, M.; Manzini, G.; De Bellis, G.; D’Alfonso, S.; et al. A Census of Tandemly Repeated Polymorphic Loci in Genic Regions Through the Comparative Integration of Human Genome Assemblies. Front. Genet. 2018, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref-Eshghi, E.; Bend, E.G.; Colaiacovo, S.; Caudle, M.; Chakrabarti, R.; Napier, M.; Brick, L.; Brady, L.; Carere, D.A.; Levy, M.A.; et al. Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions. Am. J. Hum. Genet. 2019, 104, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Shortt, J.A.; Ruggiero, R.P.; Cox, C.; Wacholder, A.C.; Pollock, D.D. Finding and extending ancient simple sequence repeat-derived regions in the human genome. Mob. DNA 2020, 11, 11–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabolacci, E.; Mancano, G.; Lanni, S.; Palumbo, F.; Goracci, M.; Ferrè, F.; Helmer-Citterich, M.; Neri, G. Genome-wide methylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in fragile X syndrome cells. Epigenetics Chromatin 2016, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Dolskiy, A.; Pustylnyak, V.; Yarushkin, A.; Lemskaya, N.; Yudkin, D.V. Inhibitors of Histone Deacetylases Are Weak Activators of the FMR1 Gene in Fragile X Syndrome Cell Lines. BioMed Res. Int. 2017, 2017, 3582601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2019, 40, 190–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Horii, T.; Hatada, I. Editing of DNA Methylation Using dCas9-Peptide Repeat and scFv-TET1 Catalytic Domain Fusions. Adv. Struct. Saf. Stud. 2018, 419–428. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Park, C.-Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.-W. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE 2016, 11, e0165499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowska, M.; Olejniczak, M. Gene Therapy for Huntington’s Disease Using Targeted Endonucleases. Methods Mol. Biol. 2020, 2056, 269–284. [Google Scholar] [PubMed]
- Zucchelli, S.; Cotella, D.; Takahashi, H.; Carrieri, C.; Cimatti, L.; Fasolo, F.; Jones, M.; Sblattero, D.; Sanges, R.; Santoro, C.; et al. SINEUPs: A new class of natural and synthetic antisense long non-coding RNAs that activate translation. RNA Boil. 2015, 12, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bon, C.; Luffarelli, R.; Russo, R.; Fortuni, S.; Pierattini, B.; Santulli, C.; Fimiani, C.; Persichetti, F.; Cotella, D.; Mallamaci, A.; et al. SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 2019, 47, 10728–10743. [Google Scholar] [CrossRef] [Green Version]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef]
- Poeta, L.; Padula, A.; Attianese, B.; Valentino, M.; Verrillo, L.; Filosa, S.; Shoubridge, C.; Barra, A.; E Schwartz, C.; Christensen, J.; et al. Histone demethylase KDM5C is a SAHA-sensitive central hub at the crossroads of transcriptional axes involved in multiple neurodevelopmental disorders. Hum. Mol. Genet. 2019, 28, 4089–4102. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poeta, L.; Drongitis, D.; Verrillo, L.; Miano, M.G. DNA Hypermethylation and Unstable Repeat Diseases: A Paradigm of Transcriptional Silencing to Decipher the Basis of Pathogenic Mechanisms. Genes 2020, 11, 684. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060684
Poeta L, Drongitis D, Verrillo L, Miano MG. DNA Hypermethylation and Unstable Repeat Diseases: A Paradigm of Transcriptional Silencing to Decipher the Basis of Pathogenic Mechanisms. Genes. 2020; 11(6):684. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060684
Chicago/Turabian StylePoeta, Loredana, Denise Drongitis, Lucia Verrillo, and Maria Giuseppina Miano. 2020. "DNA Hypermethylation and Unstable Repeat Diseases: A Paradigm of Transcriptional Silencing to Decipher the Basis of Pathogenic Mechanisms" Genes 11, no. 6: 684. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060684