Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset and Bioinformatics
2.2. Gene Set Enrichment and Modeling of Gene Interactions Networks
2.3. Statistical Analyses
3. Results
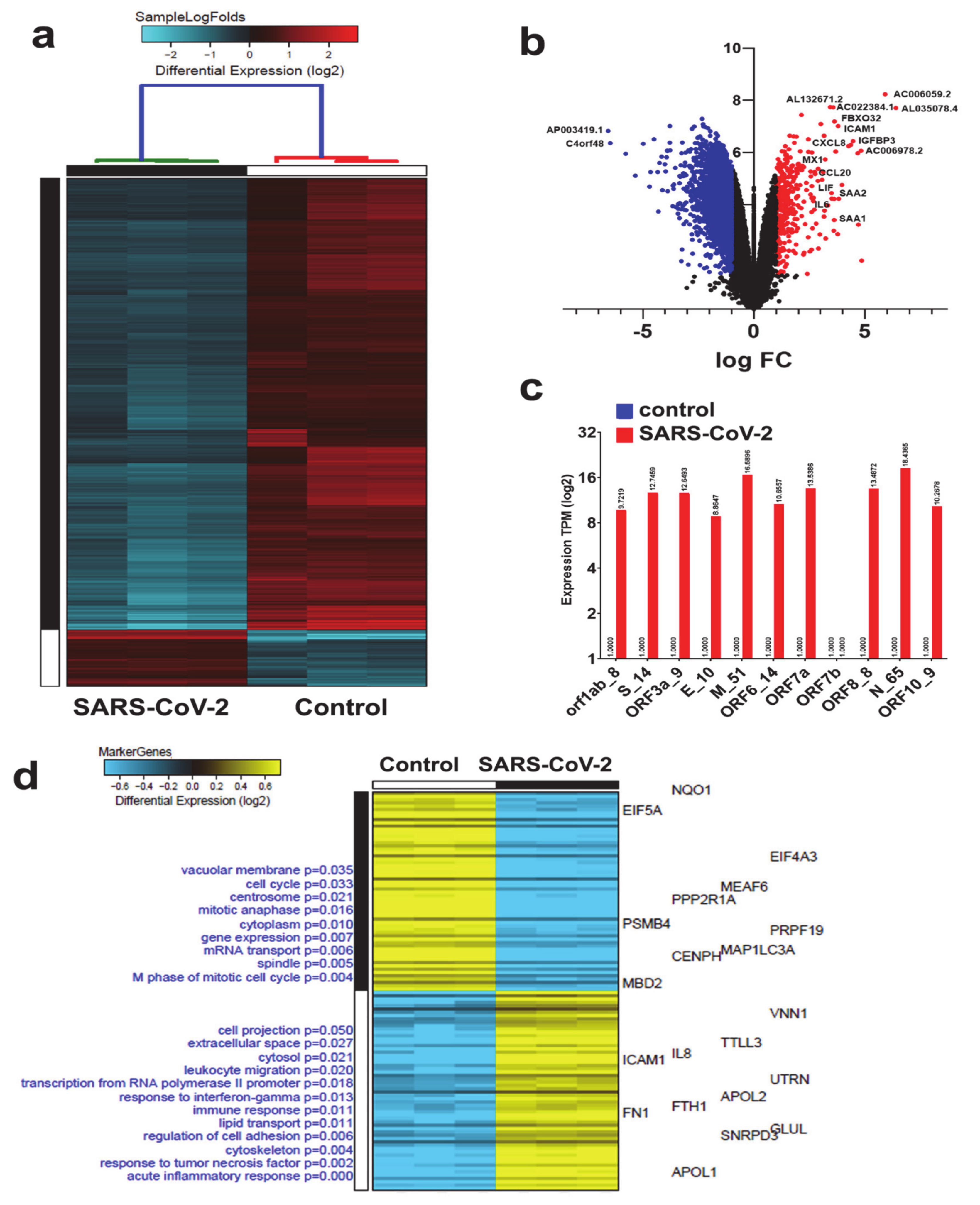
3.1. Identification of Differentially Regulated Host Cell Genes in Response to SARS-CoV-2 Infection NBHE Cells
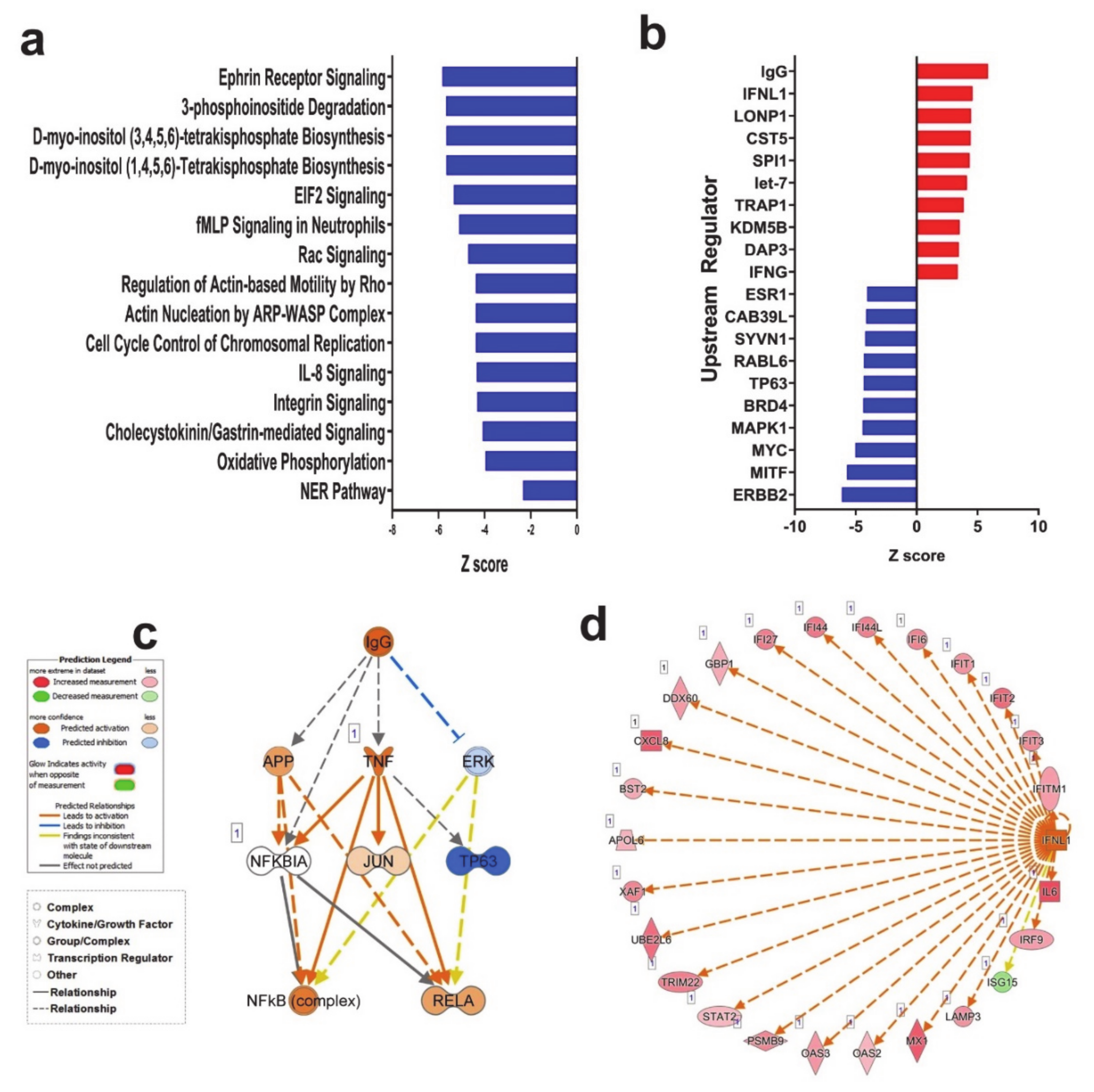
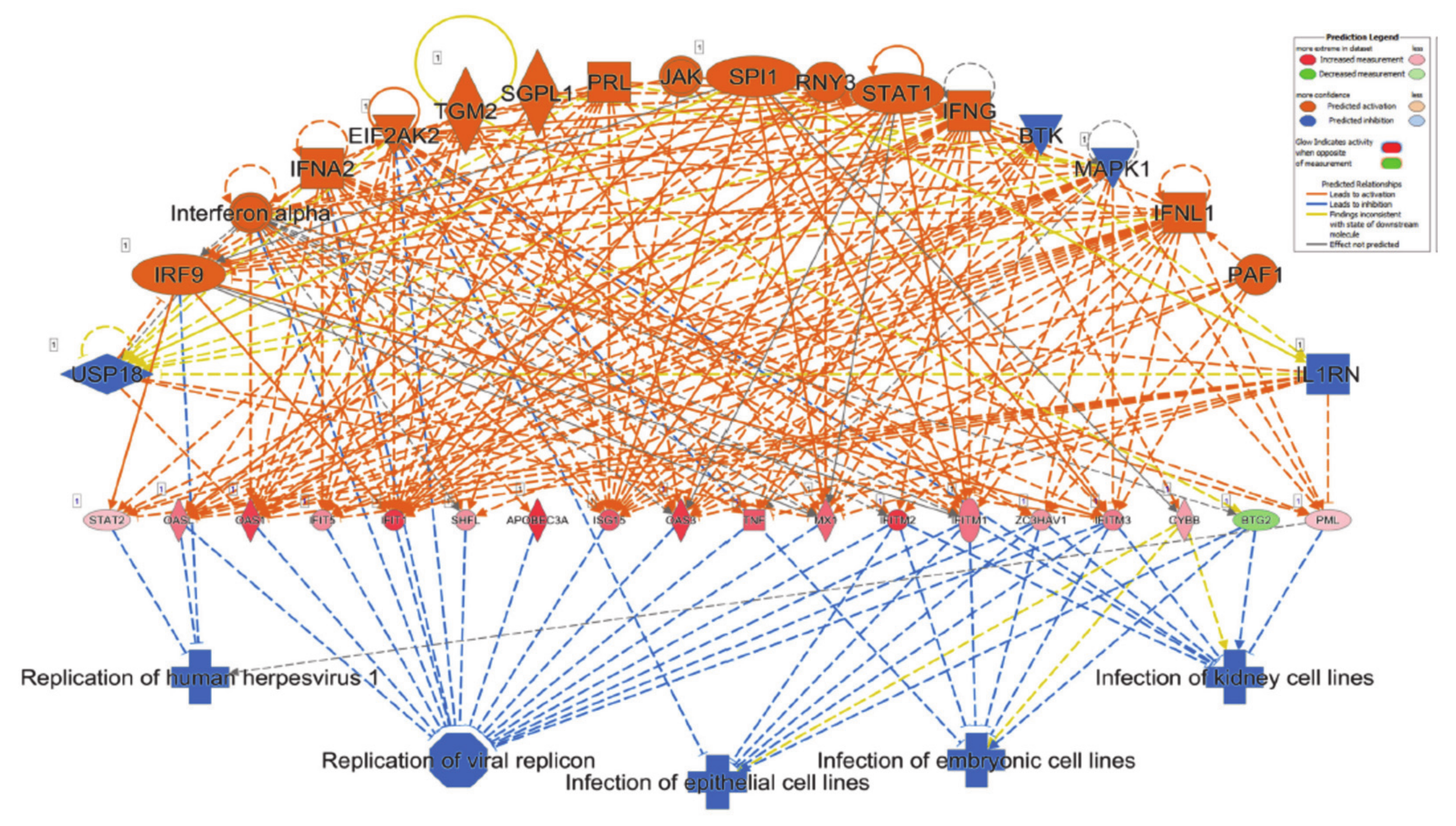
3.2. Canonical and Upstream Regulator Analysis Highlights Activation of Interferon Response Pathways in SARS-CoV-2 NHBE Cells
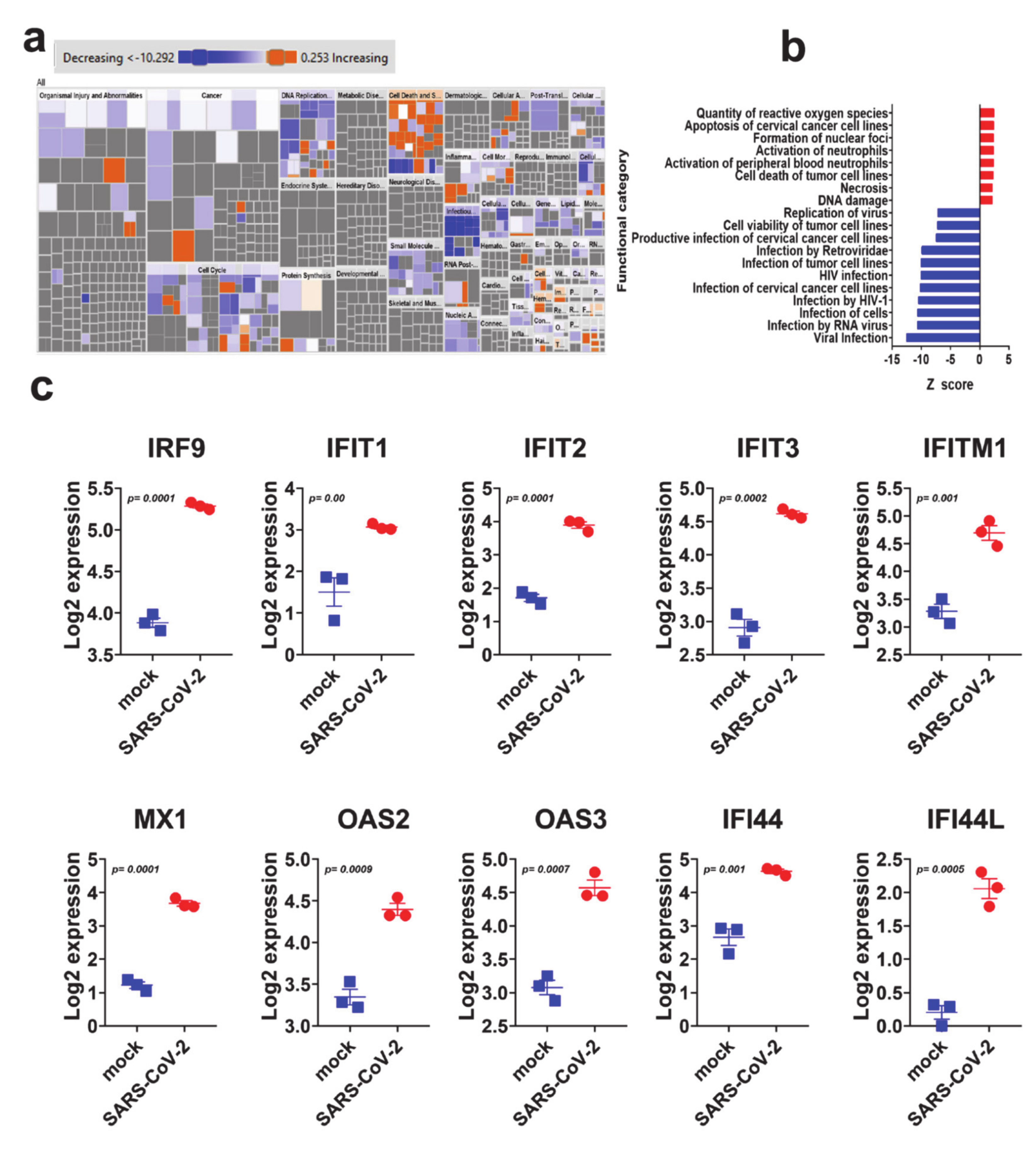
3.3. SARS-CoV-2 NHBE Gene Signature Predicted Activation of Cell Death and Inhibition of Viral Infection Based on Disease Functions Analysis by IPA
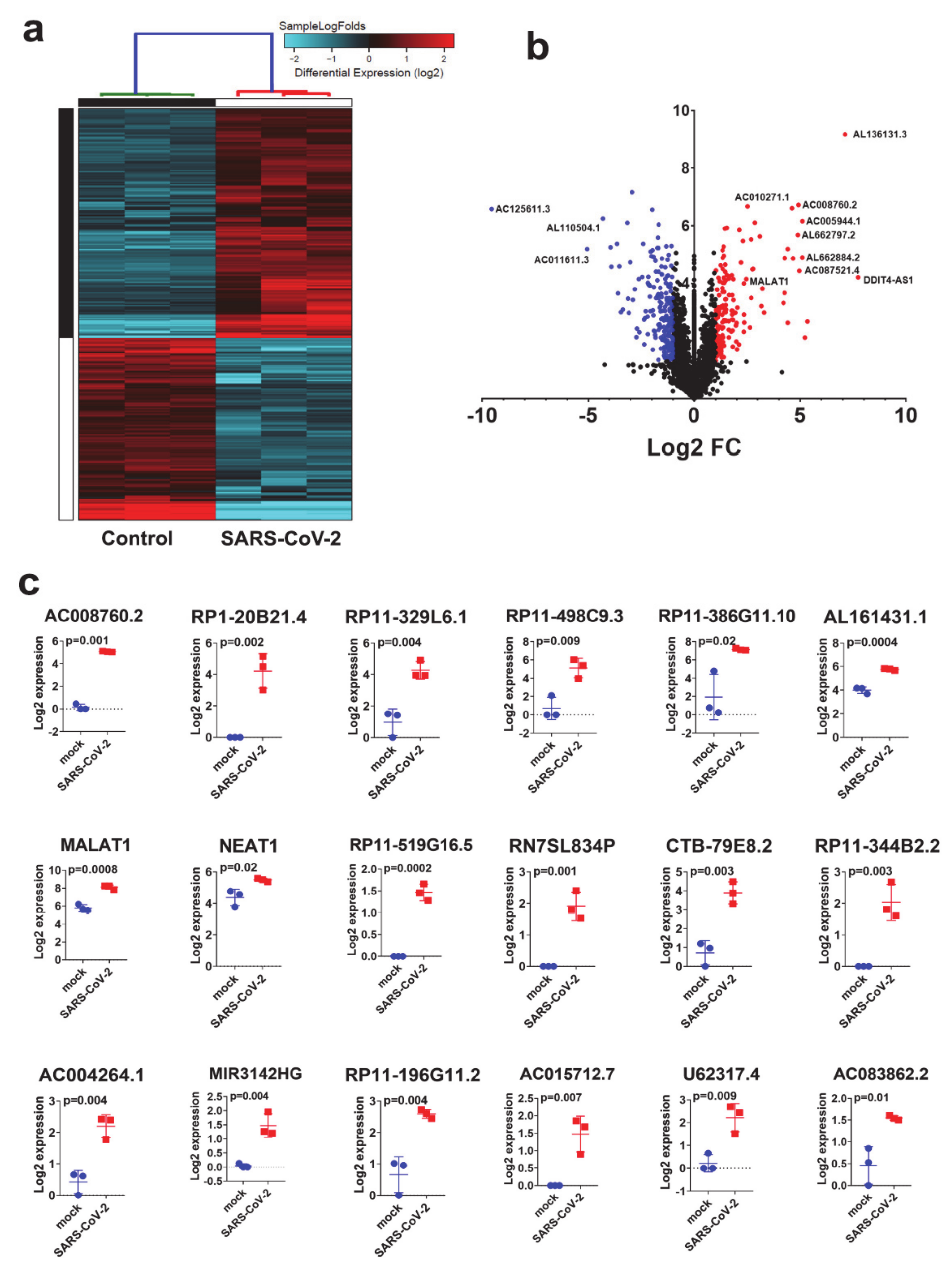
3.4. Expression Pattern of lncRNAs in SARS-CoV-2 Infected NBHE Cells
3.5. Alterations in Gene Expression and Associated Functional Classification in Lung Tissue from Patient with COVID-19 Revealed Activation of Interferon and Response to Viral Infection as the Main Hallmarks
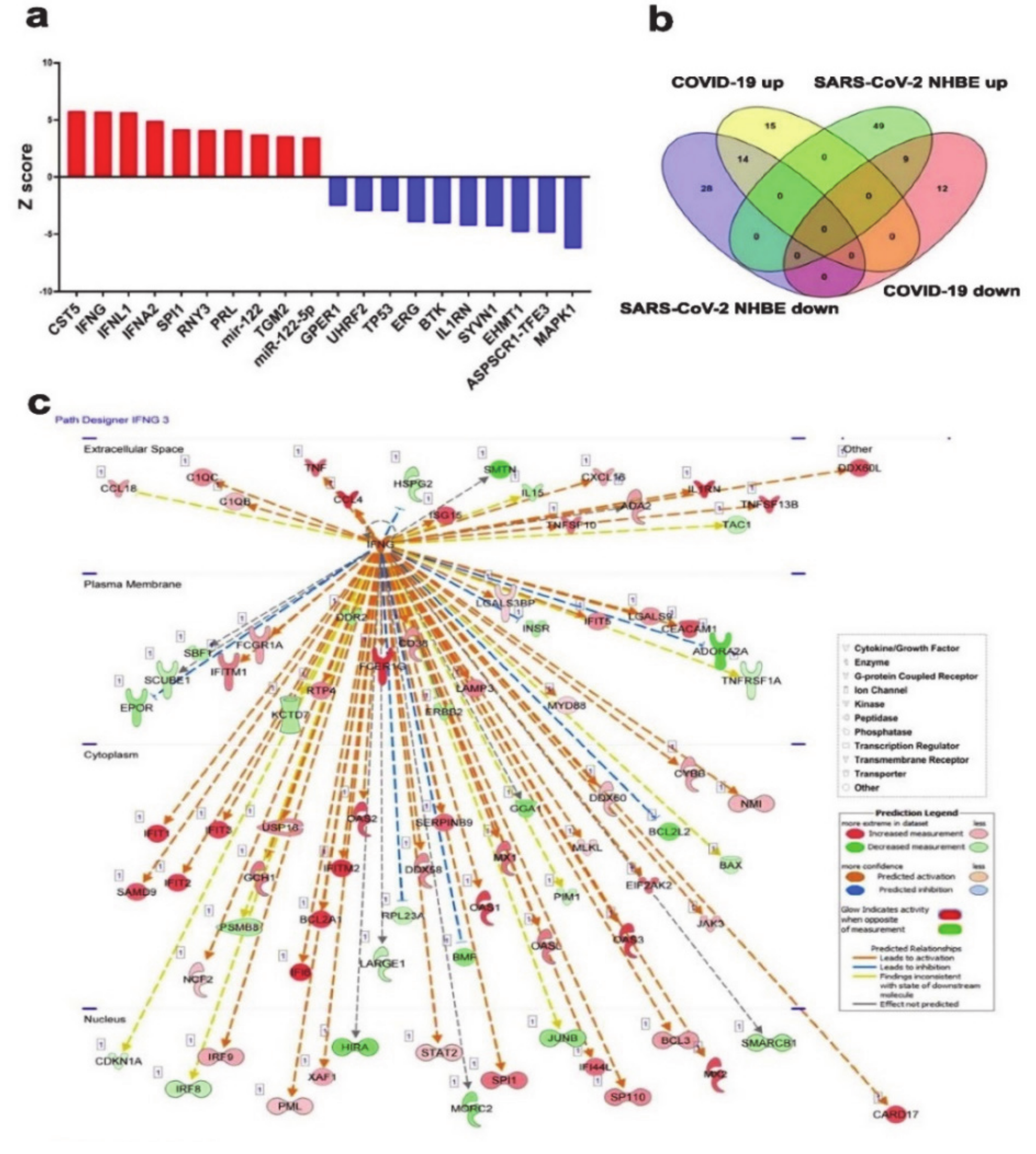
3.6. Upstream Regulator Predicts Upregulation of JAK-STAT and Interferon Cascade and miR-122 Functional Categories in Lung Tissue from COVID-19 Patient
3.7. Suppression of Viral Infection and Replication Based on Transcriptome and IPA Analysis of Lung-Derived Tissue from COVID-19 Patient
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Walls, A.C.; Tortorici, M.A.; Frenz, B.; Snijder, J.; Li, W.; Rey, F.A.; DiMaio, F.; Bosch, B.J.; Veesler, D. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2016, 23, 899–905. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease (COVID-19) Pandemic; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Hurst-Hess, K.R.; Koetzner, C.A.; Masters, P.S. Analyses of Coronavirus Assembly Interactions with Interspecies Membrane and Nucleocapsid Protein Chimeras. J. Virol. 2016, 90, 4357–4368. [Google Scholar] [CrossRef] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pohlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F. Receptor recognition mechanisms of coronaviruses: A decade of structural studies. J. Virol. 2015, 89, 1954–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012, 287, 8904–8911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.T.; Shum, M.H.; Zhu, H.C.; Tong, Y.G.; Ni, X.B.; Liao, Y.S.; Wei, W.; Cheung, W.Y.; Li, W.J.; Li, L.F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zai, J.; Zhao, Q.; Nie, Q.; Li, Y.; Foley, B.T.; Chaillon, A. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J. Med. Virol. 2020, 92, 602–611. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar] [PubMed]
- Josset, L.; Tchitchek, N.; Gralinski, L.E.; Ferris, M.T.; Eisfeld, A.J.; Green, R.R.; Thomas, M.J.; Tisoncik-Go, J.; Schroth, G.P.; Kawaoka, Y.; et al. Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol. 2014, 11, 875–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, J.T.; Varble, A.; Sachidanandam, R.; Zlatev, I.; Manoharan, M.; Garcia-Sastre, A.; tenOever, B.R. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc. Natl. Acad. Sci. USA 2010, 107, 11525–11530. [Google Scholar] [CrossRef] [Green Version]
- Bidet, K.; Dadlani, D.; Garcia-Blanco, M.A. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog 2014, 10, e1004242. [Google Scholar] [CrossRef] [Green Version]
- Morales, L.; Oliveros, J.C.; Fernandez-Delgado, R.; tenOever, B.R.; Enjuanes, L.; Sola, I. SARS-CoV-Encoded Small RNAs Contribute to Infection-Associated Lung Pathology. Cell Host Microbe 2017, 21, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Peng, X.; Gralinski, L.; Armour, C.D.; Ferris, M.T.; Thomas, M.J.; Proll, S.; Bradel-Tretheway, B.G.; Korth, M.J.; Castle, J.C.; Biery, M.C.; et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. MBio 2010, 1. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnubalaji, R.; Sasidharan Nair, V.; Ouararhni, K.; Elkord, E.; Alajez, N.M. Integrated Transcriptome and Pathway Analyses Revealed Multiple Activated Pathways in Breast Cancer. Front. Oncol. 2019, 9, 910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatasubramanian, M.; Chetal, K.; Schnell, D.; Atluri, G.; Salomonis, N. Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF. Bioinformatics 2020, 36, 3773–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaath, H.; Toor, S.M.; Nair, V.S.; Elkord, E.; Alajez, N.M. Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients. Cancers 2019, 11, 1994. [Google Scholar] [CrossRef] [Green Version]
- Zambon, A.C.; Gaj, S.; Ho, I.; Hanspers, K.; Vranizan, K.; Evelo, C.T.; Conklin, B.R.; Pico, A.R.; Salomonis, N. GO-Elite: A flexible solution for pathway and ontology over-representation. Bioinformatics 2012, 28, 2209–2210. [Google Scholar] [CrossRef] [Green Version]
- Vishnubalaji, R.; Shaath, H.; Elango, R.; Alajez, N.M. Noncoding RNAs as potential mediators of resistance to cancer immunotherapy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Testoni, B.; Vollenkle, C.; Guerrieri, F.; Gerbal-Chaloin, S.; Blandino, G.; Levrero, M. Chromatin dynamics of gene activation and repression in response to interferon alpha (IFN(alpha)) reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. J. Biol. Chem. 2011, 286, 20217–20227. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E. Whence interferon? Variety in the production of interferon in response to viral infection. J. Exp. Med. 2002, 195, F15–F18. [Google Scholar] [CrossRef] [Green Version]
- Ford, E.; Thanos, D. The transcriptional code of human IFN-beta gene expression. Biochim. Biophys. Acta 2010, 1799, 328–336. [Google Scholar] [CrossRef]
- Kroczynska, B.; Mehrotra, S.; Arslan, A.D.; Kaur, S.; Platanias, L.C. Regulation of interferon-dependent mRNA translation of target genes. J. Interferon Cytokine Res. 2014, 34, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Muendlein, H.I.; Sarhan, J.; Liu, B.C.; Connolly, W.M.; Schworer, S.A.; Smirnova, I.; Tang, A.Y.; Ilyukha, V.; Pietruska, J.; Tahmasebi, S.; et al. Constitutive Interferon Attenuates RIPK1/3-Mediated Cytokine Translation. Cell Rep. 2020, 30, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [Green Version]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway-A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef]
- Zampieri, C.A.; Fortin, J.F.; Nolan, G.P.; Nabel, G.J. The ERK mitogen-activated protein kinase pathway contributes to Ebola virus glycoprotein-induced cytotoxicity. J. Virol. 2007, 81, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Huynh, V.T.; Lim, Y.S.; Tran, S.C.; Pham, T.M.; Nguyen, L.N.; Hwang, S.B. Hepatitis C Virus Nonstructural 5A Protein Interacts with Abelson Interactor 1 and Modulates Epidermal Growth Factor-mediated MEK/ERK Signaling Pathway. J. Biol. Chem. 2016, 291, 22607–22617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Liu, J.; Liu, J. The expression of lncRNA-MALAT1 in breast cancer patients and its influences on prognosis. Cell Mol. Biol. 2020, 66, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Guo, X.; Li, Y. lncRNA MALAT1 regulates the expression level of miR-21 and interferes with the biological behavior of colon cancer cells. J. Balk. Union Ocol. 2020, 25, 907–913. [Google Scholar]
- Li, Q.; Dai, Z.; Xia, C.; Jin, L.; Chen, X. Suppression of long non-coding RNA MALAT1 inhibits survival and metastasis of esophagus cancer cells by sponging miR-1-3p/CORO1C/TPM3 axis. Mol. Cell Biochem. 2020, 470, 165–174. [Google Scholar] [CrossRef]
- Ding, H.; Wang, F.; Shi, X.; Ma, H.; Du, Y.; Hou, L.; Xing, N. LncRNA MALAT1 induces the dysfunction of beta cells via reducing the histone acetylation of the PDX-1 promoter in type 1 diabetes. Exp. Mol. Pathol. 2020, 114, 104432. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zhang, L.; Li, Z.; Xiao, D.; Zhang, Y.; Yang, H.; Liu, X.; Zhang, H.; Chen, X.; Xu, C.; et al. SIRT6-mediated transcriptional suppression of MALAT1 is a key mechanism for endothelial to mesenchymal transition. Int. J. Cardiol. 2019, 295, 7–13. [Google Scholar] [CrossRef]
- Chen, H.; Wang, X.; Yan, X.; Cheng, X.; He, X.; Zheng, W. LncRNA MALAT1 regulates sepsis-induced cardiac inflammation and dysfunction via interaction with miR-125b and p38 MAPK/NFkappaB. Int. Immunopharmacol. 2018, 55, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Li, J.; Han, Z.; Chen, Z.; Zhang, Q. Silencing of lncRNA MALAT1 Prevents Inflammatory Injury after Lung Transplant Ischemia-Reperfusion by Downregulation of IL-8 via p300. Mol. Ther. Nucleic Acids 2019, 18, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Shi, H.; Ma, N.; Zi, P.; Liu, Q.; Sun, R. BML-111 alleviates acute lung injury through regulating the expression of lncRNA MALAT1. Arch. Biochem. Biophys. 2018, 649, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Vrati, S. The Malat1 long non-coding RNA is upregulated by signalling through the PERK axis of unfolded protein response during flavivirus infection. Sci. Rep. 2015, 5, 17794. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Peng, X.; Xie, T.; Lu, X.; Liu, F.; Wu, H.; Yang, Z.; Wang, J.; Cheng, L.; Wu, N. Detection of the long noncoding RNAs nuclear-enriched autosomal transcript 1 (NEAT1) and metastasis associated lung adenocarcinoma transcript 1 in the peripheral blood of HIV-1-infected patients. HIV Med. 2016, 17, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Qu, D.; Sun, W.W.; Li, L.; Ma, L.; Sun, L.; Jin, X.; Li, T.; Hou, W.; Wang, J.H. Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter. Nucleic Acids Res. 2019, 47, 3013–3027. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chen, C.Y.; Yedavalli, V.S.; Jeang, K.T. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. MBio 2013, 4, e00596–e00512. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishnubalaji, R.; Shaath, H.; Alajez, N.M. Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response. Genes 2020, 11, 760. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070760
Vishnubalaji R, Shaath H, Alajez NM. Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response. Genes. 2020; 11(7):760. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070760
Chicago/Turabian StyleVishnubalaji, Radhakrishnan, Hibah Shaath, and Nehad M. Alajez. 2020. "Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response" Genes 11, no. 7: 760. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070760