1. Introduction
DNA methylation has been found a promising biomarker in cancer detection and cancer classification. DNA methylation can be defined as a heritable epigenetic mark where a methyl group can transfer covalently to the C-5 position of the cytosine ring of DNA through DNA methyltransferases (DNMTs). DNA methylation is vital for normal development. It plays very important role in a number of key operations including genomic imprinting, inactivation of X-chromosome, repression of repetitive element transcription and transposition, and different diseases including cancer [
1]. To biologically interpret the DNA methylation data, two kinds of analysis are available: (i) single differentially methylated genes (CpG sites) finding [
2,
3] and (ii) differentially methylated region (DMR) finding [
4,
5,
6]. These two kinds of analysis are only specific to performing a single task. Therefore, it is important to incorporate different factors to correctly interpret DNA methylation data by which it can work as multi-functionalities from different directions such as prediction of gene expression using DNA methylation, differential expression analysis, cancer classification [
7], hub gene finding, and others.
In practical scenarios, it is observed that DNA methylation normally reduces gene expression levels [
8,
9]. However, this opinion varies on different factors. There are different kinds of method to integrate DNA methylation and gene expression data. There are several shortcomings of those existing methods. Firstly, it is not easy to determine the directionality of the evaluated gene expression estimated from the DNA methylation. Normally, the suppression of gene expression is caused by hypermethylation in the promoter region, while the activation correlates the hypermethylation in the gene body. Therefore, the prediction of changing in gene expression based on simple DNA methylation results is difficult [
10]. Secondly, an accurate measure of gene promoter methylation is difficult due to the variance in the size of canonical promoters as well as the presence of the distal augments, which initiates biases into the association of methylated regions with gene models [
10]. Thirdly, the high probability of selecting a long gene due to the nearby differentially methylated CpGs or overlapping (or nested) with other genes [
10]. Fourthly, some specific tools are required for reformatting the methylation data into the genomic region formats (e.g., BED) for some web-based methods such as GREAT [
11], Galaxy [
12]. It creates more complications in their usage [
10].
Cervical cancer is a cancer which starts in the cervix, a hollow cylinder that connects the lower part of uterus to a woman’s vagina. Most of the cervical cancers grow in the cells on the outer surface of the cervix. Normally women are unable to realize this disease in the initial stage since the symptoms are more or less similar with the common conditions such as menstrual periods and urinary tract infections. The normal symptoms of the cervical cancer include abnormal bleeding during mensuration time or after having sex, pain in the pelvis, as well as pain during the urination [
13]. Here, we used a DNA methylation dataset for uterine cervical cancer from NCBI (Accession ID: GSE30760) [
14] which have two types of samples, one is normal sample and another one is uterine cervical cancer sample.
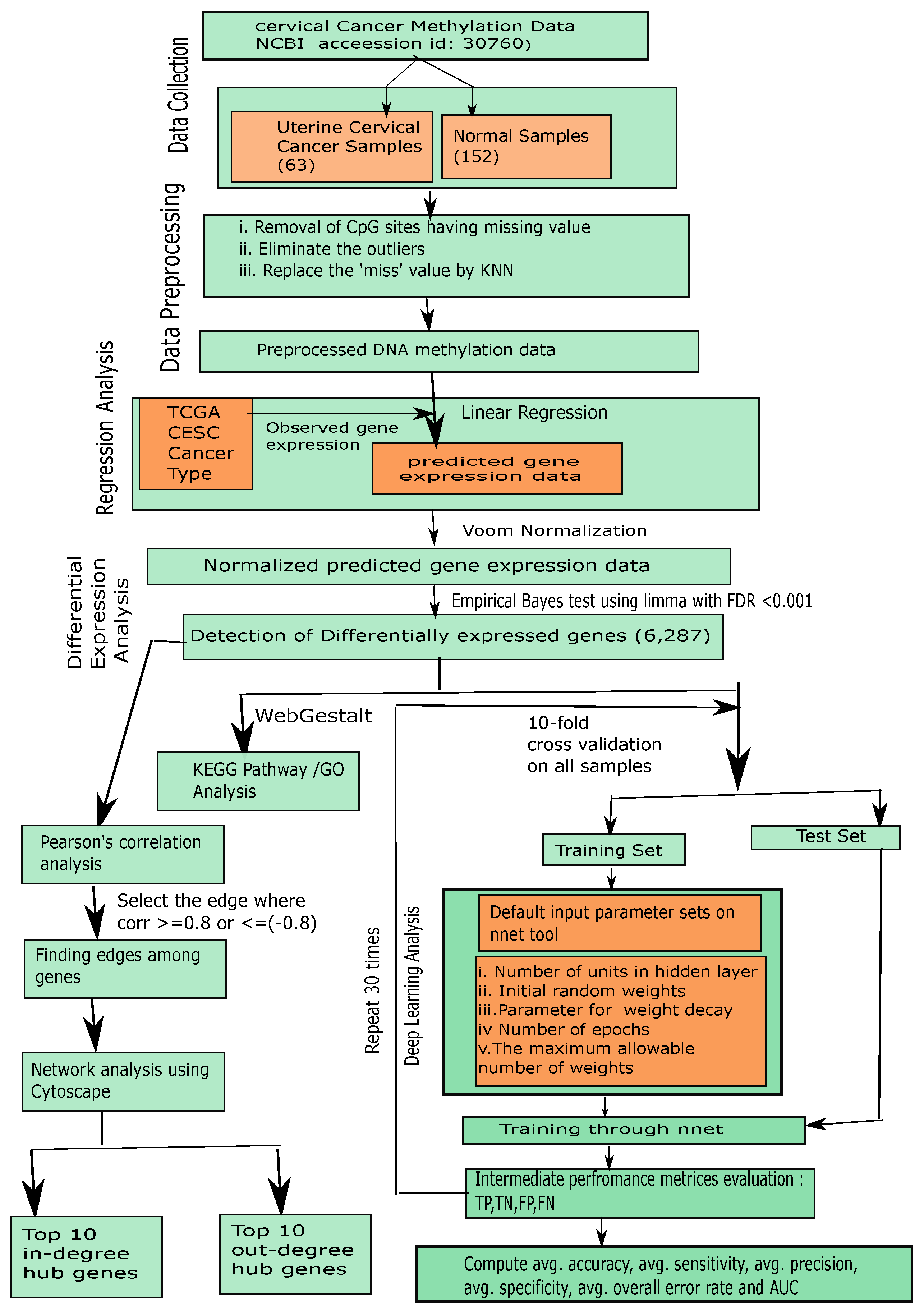
So far, there has been no method to integrate regression, differential expression and deep learning strategies for accurate interpretation of DNA methylation in a complex disease like cancer. To resolve the previously mentioned drawbacks, in this article, we provided an integrated framework using regression, differential expression and deep learning methods to correctly interpret biologically of the DNA methylation data through integrating that DNA methylation data and corresponding TCGA (The Cancer Genome Atlas) gene expression data for uterine cervical data (NCBI accession ID GSE30760) [
14,
15,
16]. We pre-filtered the redundant CpG sites, eliminated outliers, and replaced missing values. Next, we predicted corresponding gene expression value from the pre-filtered DNA methylation data through linear regression algorithm where the impact between DNA methylation and TCGA gene expression has been determined. As a result, we obtained the predicted gene expression matrix for the preprocessed DNA methylation data. Through the entire analysis, we used ByMethyl R package [
10]. Next, we identified differentially expressed genes (DEGs) using downstream analysis, Empirical Bayes test using
[
17,
18,
19]. After we applied a recently released deep learning method, “
nnet” (feed-forward neural network based model) [
20] to interpret those DEGs for determining the classification capacity of uterine cancer and normal groups, we then estimated all classification metrics (average accuracy, average sensitivity, average specificity, average precision, average overall error rate and area under curve (AUC)) using 10-fold cross validation. We trained our predicted DEG expression data using “
nnet” with the default parameter settings (i) size (=number of units in hidden layer), (ii) rang (=initial random weights) while [−rang, rang], (iii) decay (=parameter for weight decay), (iv) maxit (=the maximum number of iterations or number of epochs), (v) MaxNWts (=the maximum allowable number of weights) and other default parameters. Remarkably, we obtained
(
) as average classification accuracy of the uterine cervical cancer samples and normal samples by using DEG expression data. According to comparative study, the classification accuracy of our proposed method is higher than that of other state-of-the-art methods. We further performed in-degree and out-degree hub gene network analysis using
[
21]. We reported the five top in-degree genes (
,
,
,
and
) and the five top out-degree genes (
,
,
,
and
). After that, we performed Gene Set Enrichment Analysis (GESA) to determine enriched KEGG pathways and Gene Ontology (GO) terms including Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) on the set of all DEGs having
using
WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) [
22]. Finally, our proposed integrated framework using linear regression, differential expression and deep learning method can interpret the DNA methylation data better than using single differential methylation analysis or differentially methylated region finding strategies for any kind of cancer.
3. Results and Discussion
In this case study, we had 27,578 features and 215 samples including 152 normal samples and 63 uterine cervical cancer samples. After data preprocessing, linear regression and differential expression analysis, we obtained 6287 DEGs having
by
, in a list accompanied by computed
t-score,
p-value and FDR. Top 25 DEGs are shown in
Table 1. For example,
was the topmost DEG with minimum FDR (FDR =
). We provided the list of all DEGs obtained by differential expression analysis by Empirical Bayes test using
with FDR corrected
p-value in a
Supplementary File, Additional file 1:
Table S1. Furthermore, the predicted gene expression matrix of all DEGs computed from original pre-filtered uterine cervical cancer DNA methylation data through linear regression analysis was provided in another
Supplementary File, Additional file 2:
Table S2.
After that, we applied the latest deep learning method “
nnet” (feed-forward neural network based model), [
20] on our computed DEG expression dataset which have 6287 features with 215 samples. We used this deep learning technique with 10-fold cross validation to examine the class-label (normal and uterine cervical cancer groups) of the differentially expressed genes with a repeat of 30 times. In the cross-validation, we divided all the samples of the predicted gene expression data of the DEGs into 10 folds of samples of which nine-fold of samples was used as training set, while the remaining one-fold of the samples was utilized as the test set. From this sub step, we ran “
nnet” tool using maxit (number of epochs) equal to 100, that means the deep learning method was internally repeated for 100 times, and then computed the classification metrics at one time iteration of each fold. From this sub step, we obtained a confusion matrix consisting of True Positive (TP), False Negative (FN), False Positive (FP) and True Negative (TN). This sub procedure was repeated for each fold of samples (i.e., nine other folds). Then, we added all these metrics for these 10 times internal repetitions and produced a final confusion matrix. Thereafter, we repeated this entire procedure for multiple times (30 times) and obtained thirty confusion metrics. Using this, we obtained the average classification metric values (average accuracy, average sensitivity, average specificity, average precision, average overall error rate and area under curve (AUC)). Note that our deep learning method has already repeated 30,000 times (
) from which we computed the average accuracy, where every sample was used as a test set at least once (i.e., no sample was missing as a test sample). Here we used test sample as validation 163 sample. In this deep learning method, we used “
nnet” with the default parameter settings (i) size (=number of units in hidden layer) (=2), (ii) rang (=initial random weights)(=0.1) while [−rang, rang], (iii) decay (=parameter for weight decay)(=
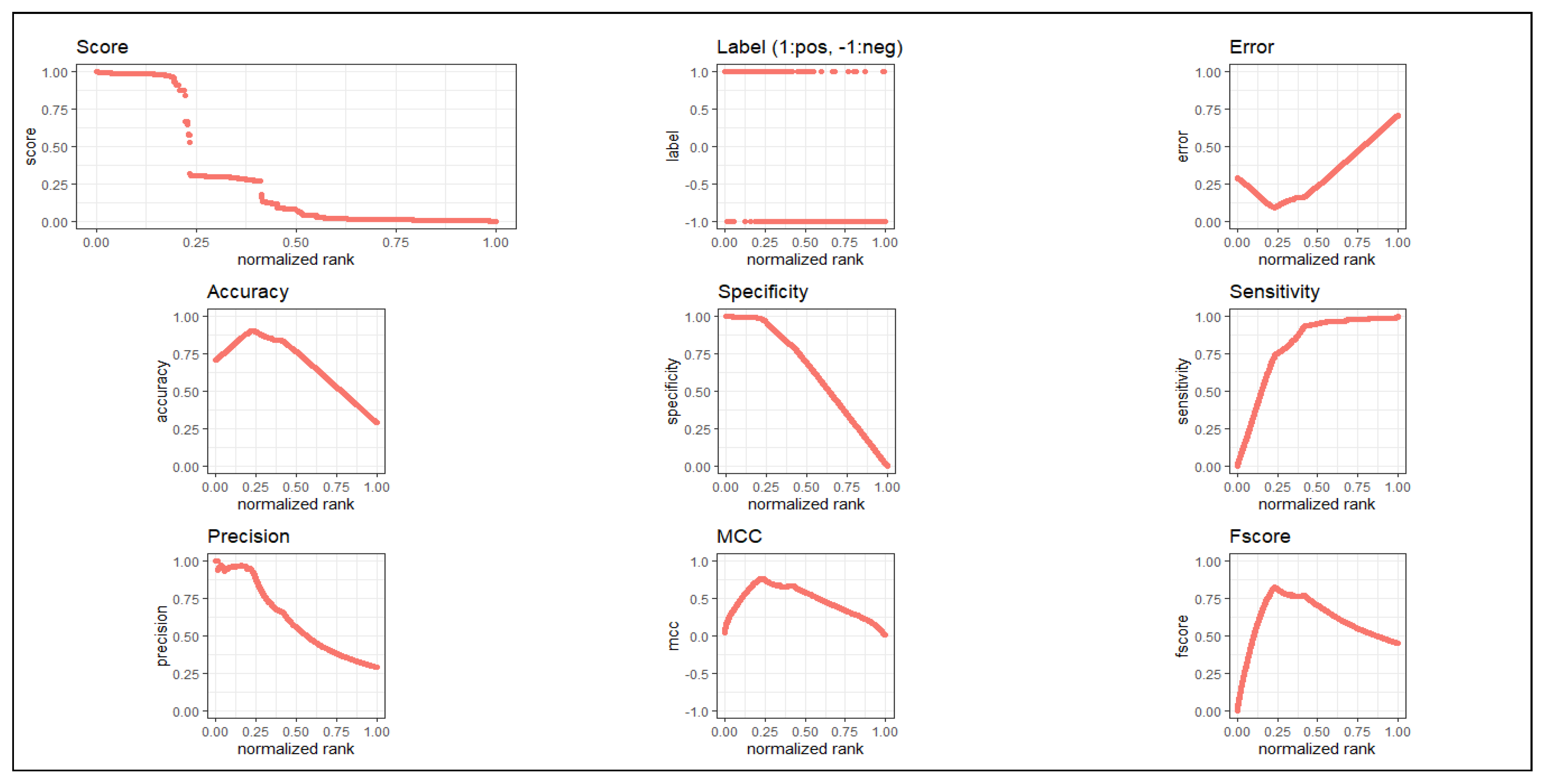
), (iv) maxit (=the maximum number of iterations or number of epochs)(=100), (v) MaxNWts (=the maximum allowable number of weights)(=84,581) and other default parameters. As we used 10-fold cross validation, 9/10 of 215 samples (i.e., 194 or 193 samples) were considered as training set and 1/10 of 215 samples (i.e., 21 or 22 samples) were taken as test set. of which nine-fold of samples was used as a training set, while remaining one-fold of samples was utilized as a test set. Thus, each sample participated in each role, either in training sample or test sample, at least once. Here, we also used the test sample as the validation sample. We obtained
(
) average classification accuracy and value of AUC was 0.858. For more details, see
Table 2. We have plotted all metrics in
Figure 2.
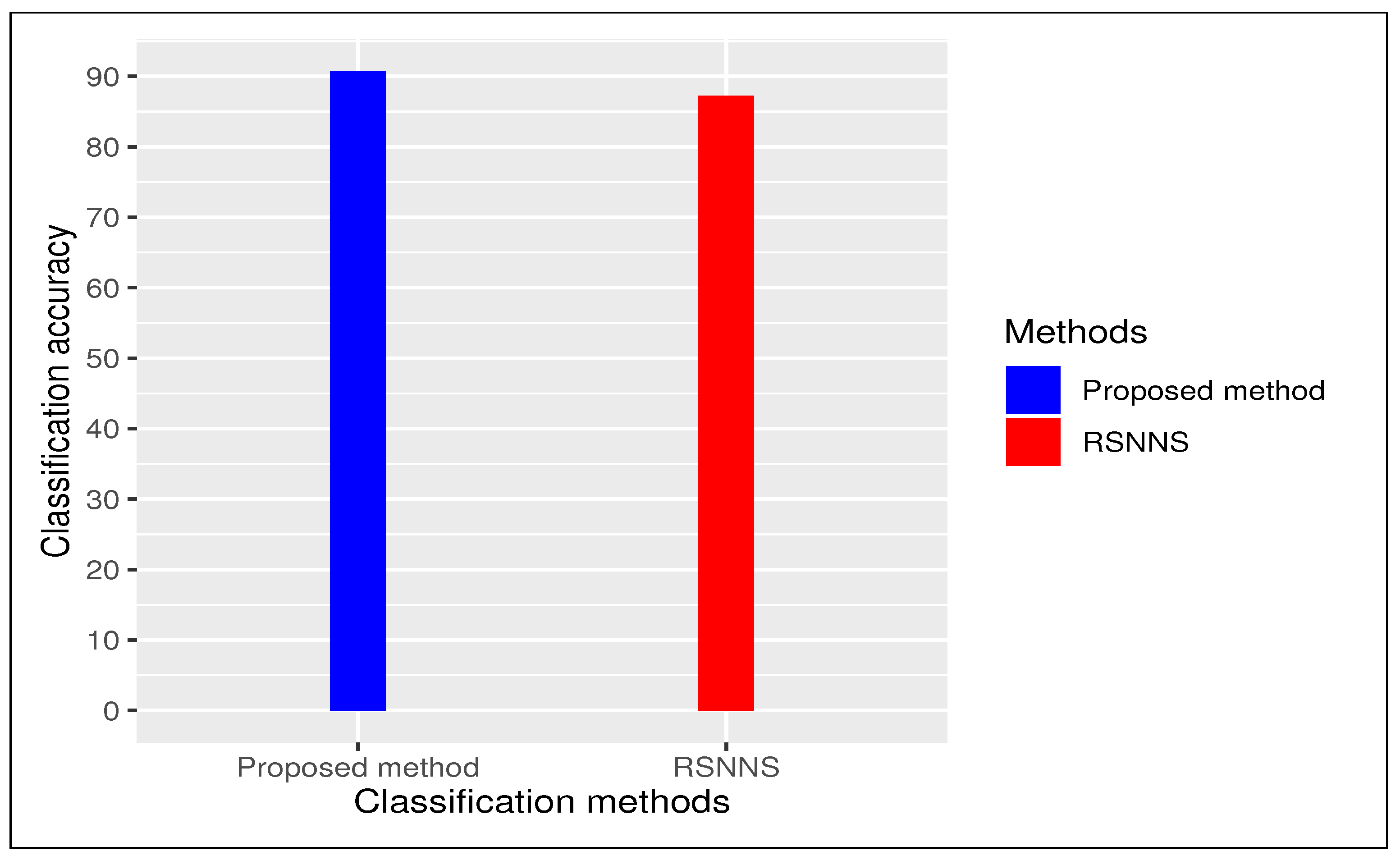
We carried out a comparative study between proposed method and an existing method “
RSNNS” (Stuttgart Neural Network Simulator (SNNS) based deep learning tool) with 10-fold cross validation with repeating 30 times. In case of “
RSNNS” we also used same default parameter settings like (i) size (=number of units in hidden layer)(=2), (ii) maxit (=maximum number of iterations or number of epochs) (=100), among others. In both cases we have repeated entire procedure 30 times to to obtain a reliable classification. Our proposed method produced an average classification accuracy of
(
) whereas existing method “
RSNNS” had
(
) as average classification accuracy (see
Figure 3). We considered our framework had better performance than all other methods using deep learning tool.
Here, we applied Pearson’s correlation analysis on our DEGs for finding out edges among genes having correlation value greater than or equal to 0.8 or, less than or equal to (−0.8). Then, we also performed in-degree and out-degree hub gene network analysis using
[
21]. As an example the five top genes with highest in-degree values were namely
,
,
,
and
, see
Table 3. Similarly, the five top most out-degree genes were namely
,
,
,
and
, see
Table 4. We provided detail hub gene network structure in a
Supplementary File, Additional file 7:
Table S7.
In the corresponding literature survey, we found that most of the topmost hub genes detected by our method played an important role in the respective cancer.
gene and cervical cancer were found to be associated by Berlanga et al. [
26].
was utilized as the negatively regulator of p53 in tumorigenesis [
27]. It had been also used as a potential bio-marker in DNA methylation at the time of treatment and risk assessment of cancer. Methylation of
might be a protective factor in the development of tumor [
28].
gene and cervical cancer were reported in the literature Broniarczyk et al. [
29]. Similarly,
gene is involved in cervical cancer, as reported in Sundaram et al. [
30], while
gene was associated with cervical cancer in Feron et al. [
31]. Similarly, the topmost out-degree hub genes were mostly associated with cervical cancer through literature search. For example, the association between
and cervical cancer were documented in Wen et al. [
32], whereas
was connected with the respective cervical cancer in Luo et al. [
33]. In addition,
and cervical cancer are reported in Liang et al. [
34], while
was found to be linked to cervical cancer in Zhang et al. [
35].
These 6287 DEGs, which have
, were taken for Gene Set Enrichment Analysis using
WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) [
22]. We had applied
WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) database on our DEG set to obtain all KEGG pathways and Gene Ontology (GO) terms [Biological Process (BP), Cellular Component (CC) and Molecular Function (MF)], accompanied by number of genes in that pathway or GO-term, enriched
p-value and FDR. Here, we took our input data set in the prescribed format of WebGestalt which was in a two-columns pattern, first one was gene name and second one was score. Here we used
t-score as score. Significant pathways and GO-terms were described in below and also for more details see
Table 5,
Table 6,
Table 7 and
Table 8. For example,
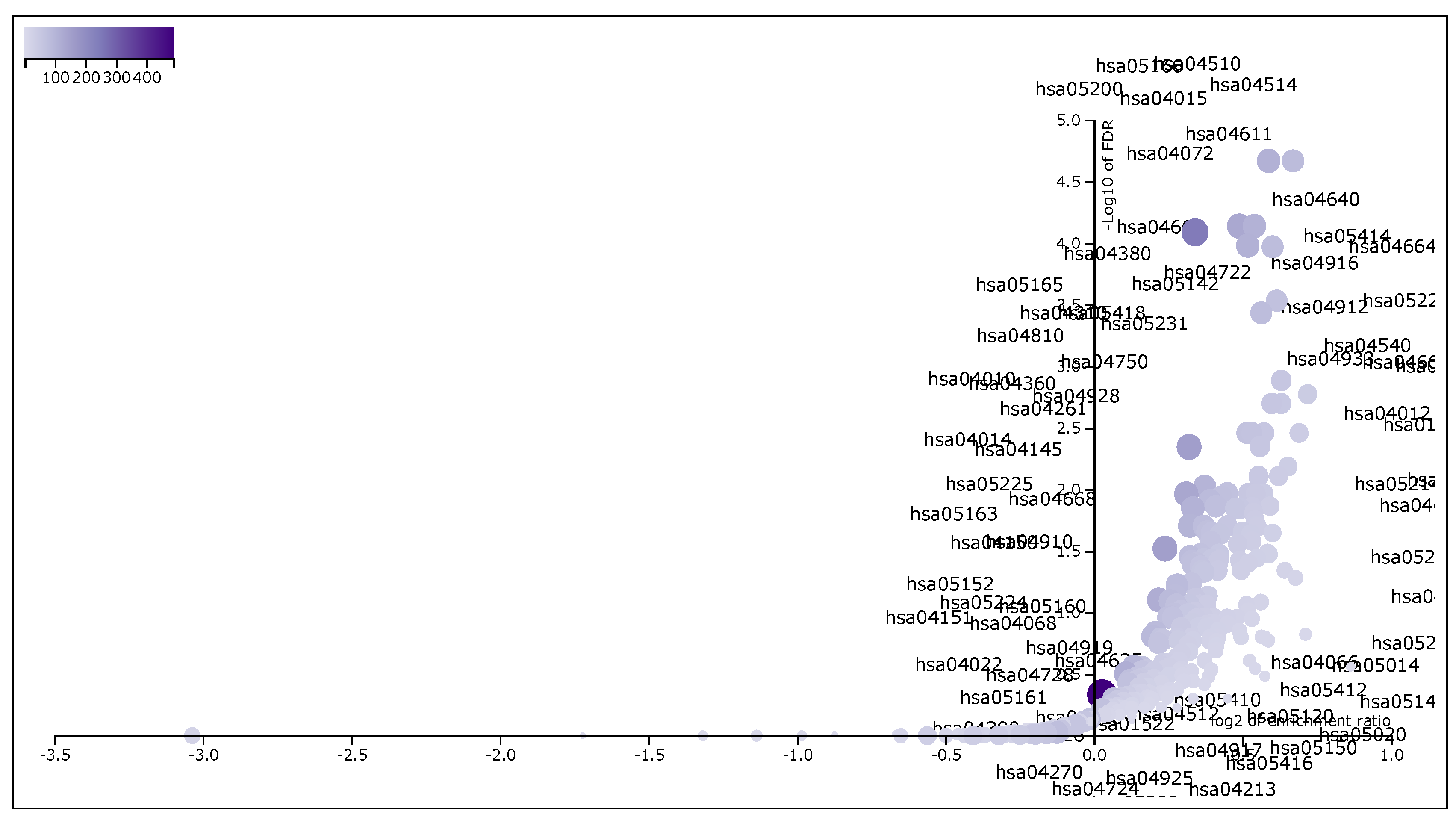
hsa05205:Proteoglycans in cancer was a top significant KEGG pathway which has minimum FDR value (
). A total of 198 genes were associated in this pathway with enriched
p-value
. For the remaining top 10 significant KEGG pathways, see
Table 5. We provided the list of all KEGG pathways in a
Supplementary File, Additional file 3:
Table S3. In addition, the volcano plot of the of normalized enrichment score of those FDR significant KEGG pathways is shown in
Figure 4. Similarly,
GO:0008283 cell proliferation was one of the top significant GO-BP terms with FDR value 0. A total of 1986 genes were associated with this GO-BP term, enriched p-value 0. For the remaining terms, see
Table 6. We provided the list of all GO-BP terms in a
Supplementary File, Additional file 4:
Table S4. In such analysis, we found
GO:0005783 endoplasmic reticulum as one of the top significant GO-CC terms with FDR value 0. A total of 1861 genes were associated with this GO-CC term, enriched
p-value 0. For the rest, see
Table 7. We provided the list of all GO-CC terms in a
Supplementary File, Additional file 5:
Table S5. Furthermore,
GO:0042802 identical protein binding was one of the top significant GO-MF terms with minimum FDR value 0. A total of 1696 genes were associated with this GO-MF term having the enriched
p-value 0. For details, see
Table 8. We provided the list of all GO-MF terms in a
Supplementary File, Additional file 6:
Table S6.
4. Conclusions and Future Work
In this article, we provided a framework using linear regression, differential expression, and deep learning to provide correct biological interface for integrating DNA methylation and corresponding TCGA gene expression data to uterine cervical cancer. To develop the framework, first we eliminated outliers, then applied linear regression to determine predicted gene expression data from the preprocessed DNA methylation data by the use of TCGA gene expression data. Then we identified 6287 differentially expressed gene with FDR cut off less than 0.001 using downstream analysis through Empirical Bayes test using . After that, we applied “nnet” deep learning method to interpret differentially expressed genes with 10-fold cross validation and with the default parameter settings (i) size (=number of units in hidden layer), (ii) rang (=initial random weights) while [−rang, rang], (iii) decay (=parameter for weight decay), (iv) maxit (=the maximum number of iterations or number of epochs), (v) MaxNWts (=the maximum allowable number of weights) and other default parameters also. We obtained () as average classification accuracy of the uterine cervical cancer samples and normal samples for DEG expression data, which is more significant than other existing methods. So through the deep learning and comparative study, we can say that our obtained DEGs are strong and efficient in disease classification.
Here, we also performed in-degree and out-degree hub gene network analysis using
[
21]. We reported the five highest in-degree genes (
,
,
,
and
) and the five highest out-degree genes (
,
,
,
and
). Furthermore, we used pathway analysis on DEGs with
using
. Finally, our framework is useful for better biological interpretation of the DNA methylation data rather than single differential methylation analysis or differentially methylated region finding.
In our future study, we will extend our current work through integrating random forest ensemble method into deep learning strategy to obtain a better classification model in all prospective, and then apply that on big data (e.g., single cell RNA sequencing data or, other TCGA cancer tissue specific data) for cancer classification.







{kind=link}
{kind=link}
{kind=link}
{kind=link}