Stable Longitudinal Methylation Levels at the CpG Sites Flanking the CTG Repeat of DMPK in Patients with Myotonic Dystrophy Type 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Material
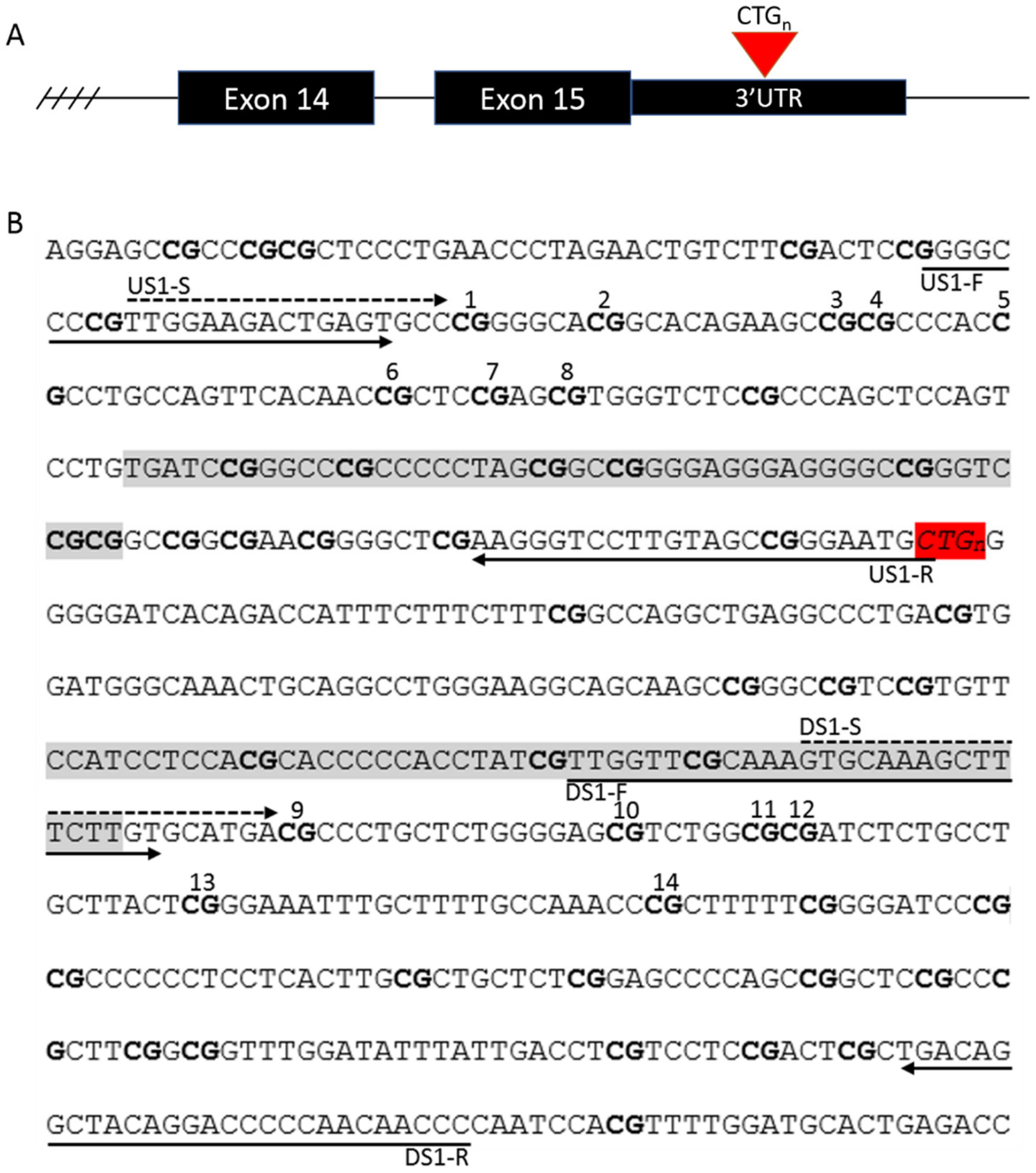
2.2. Assessment of Repeat Interruptions
2.3. DNA Extraction and Methylation Analysis
2.4. Statistical Analysis
3. Results
3.1. Repeat Interruptions
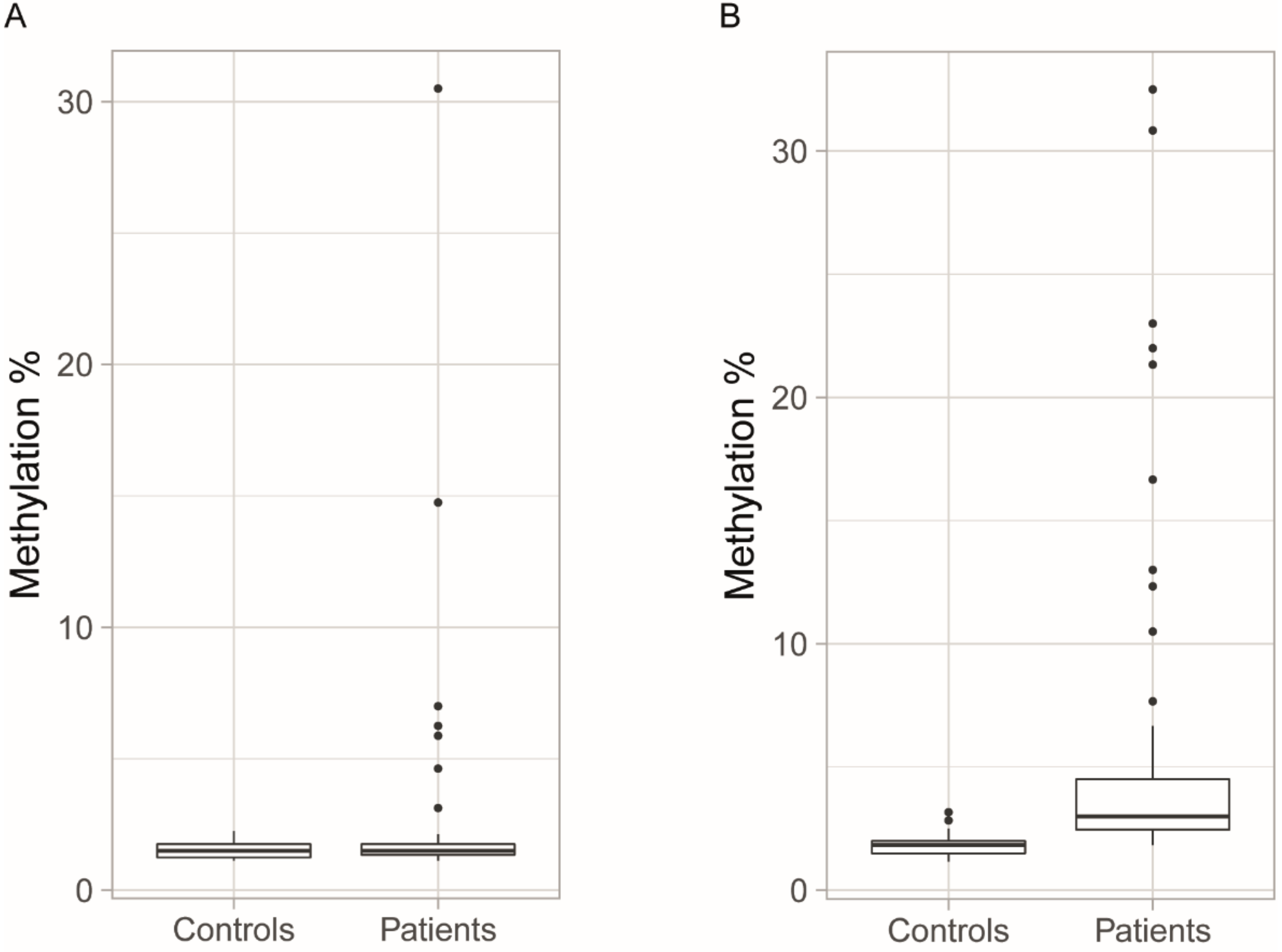
3.2. Methylation Levels in Initial Blood Samples
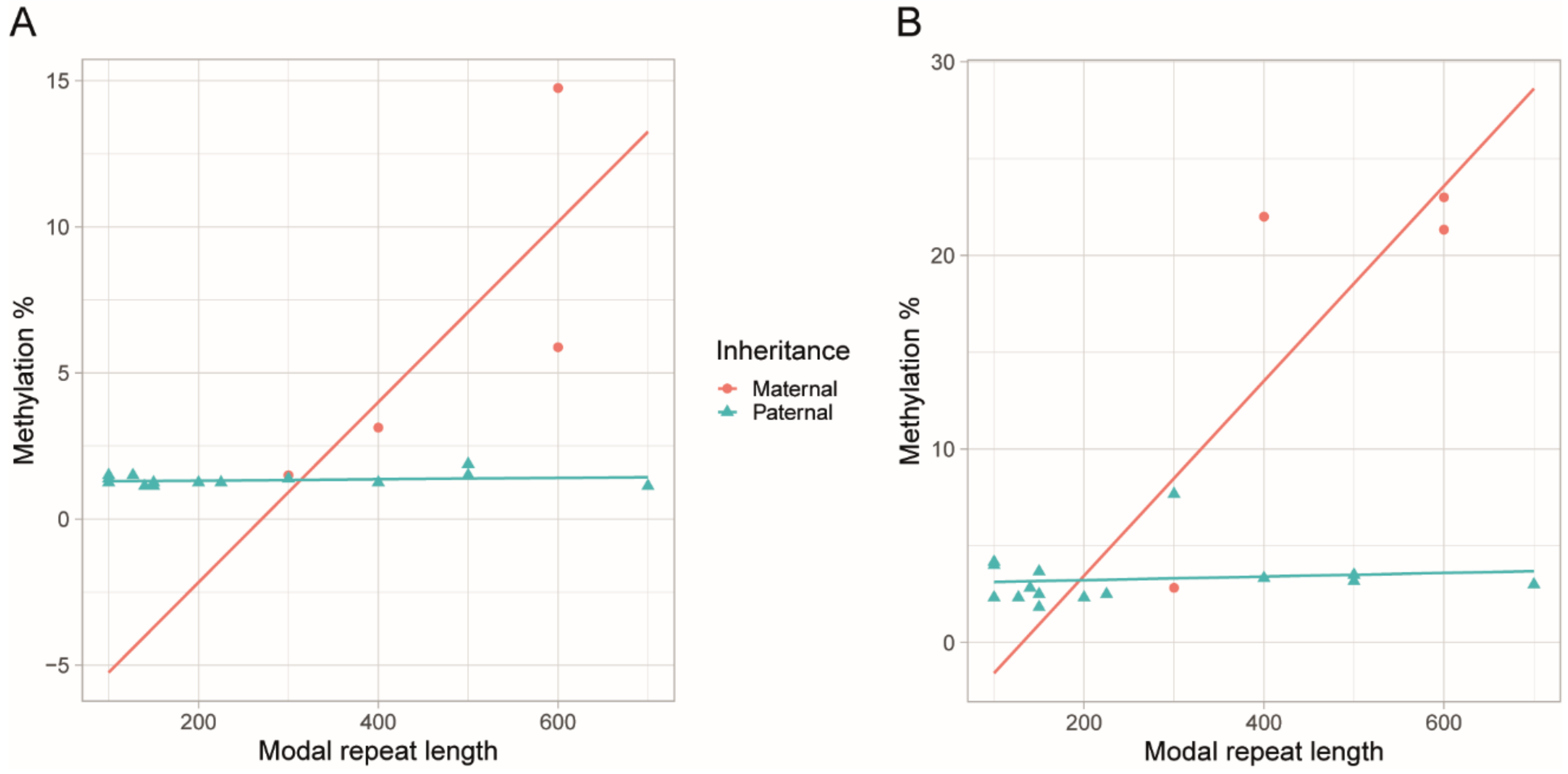
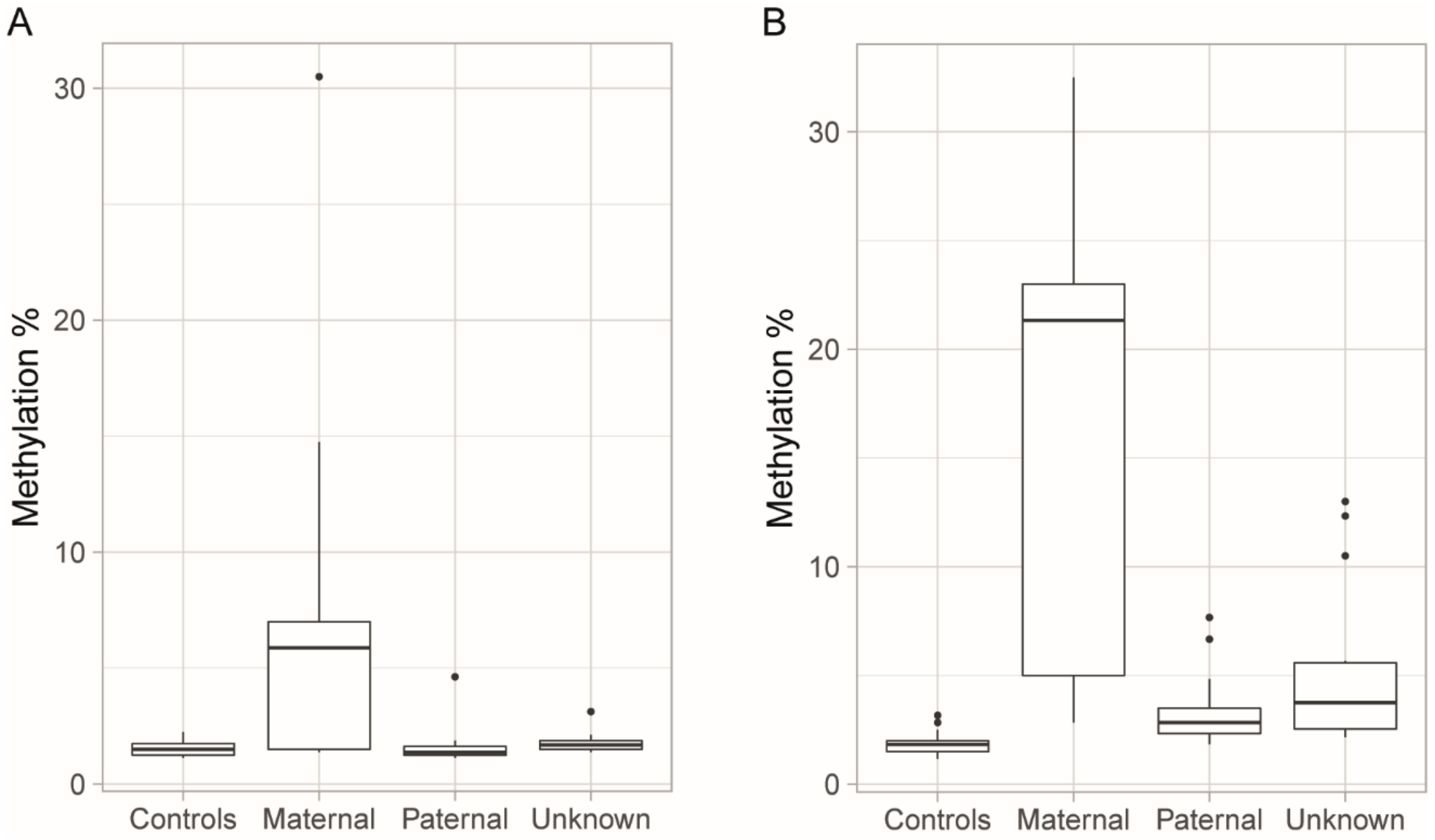
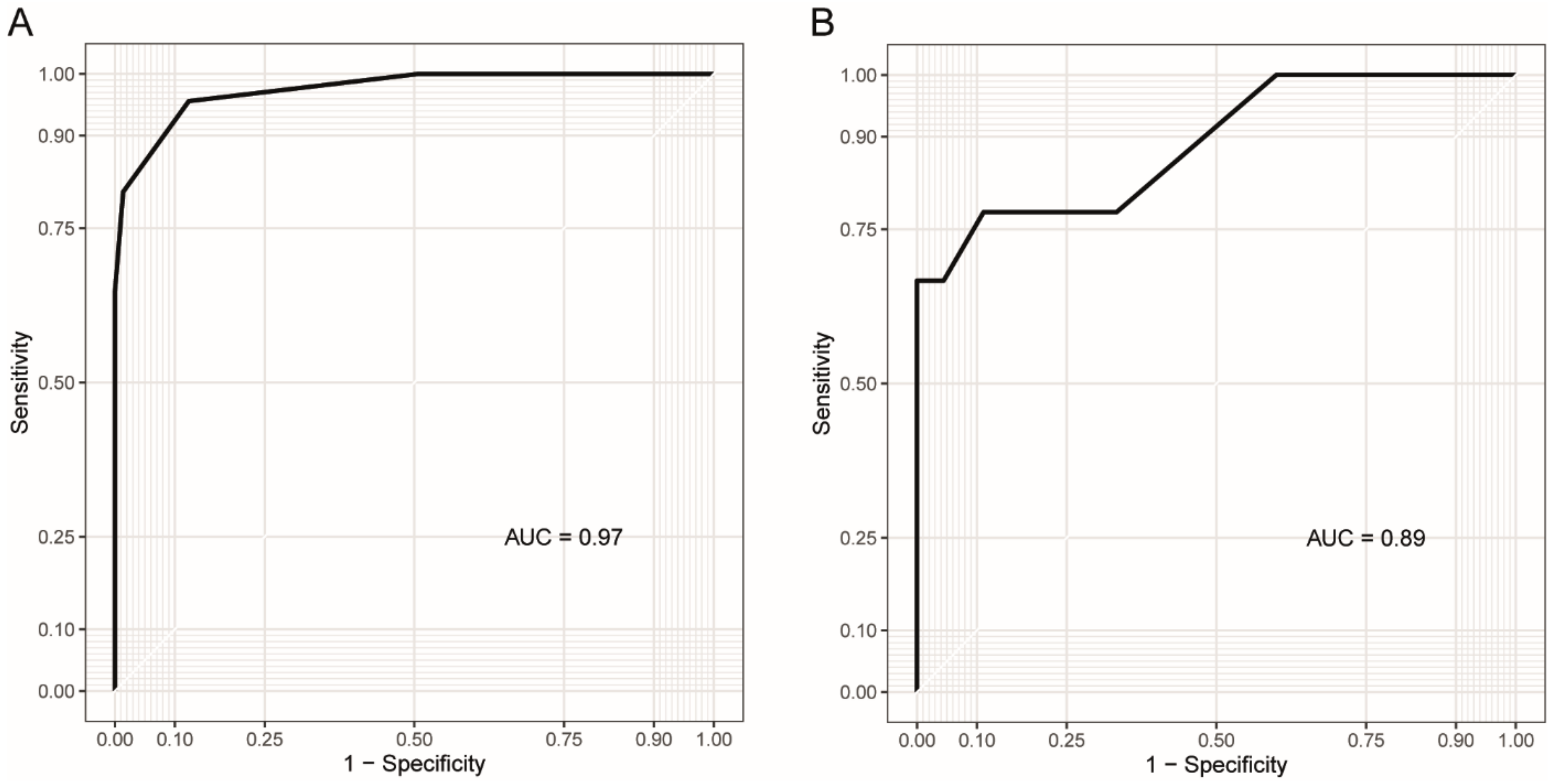
3.3. Blood Methylation as a Biomarker for DM1 and Disease Inheritance
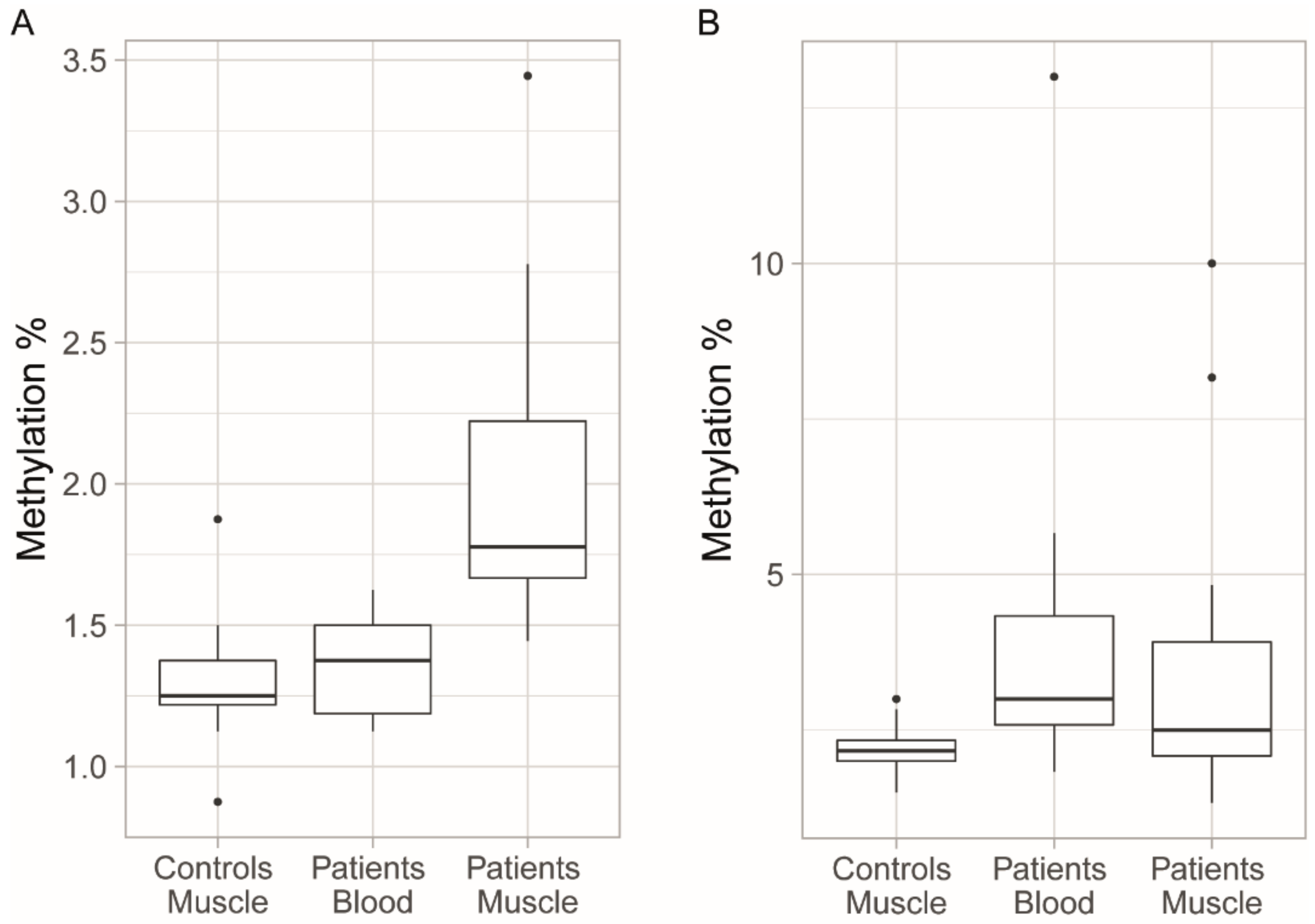
3.4. Methylation Levels in Muscle
3.5. Longitudinal Changes in Methylation Levels in Blood
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schara, U.; Schoser, B.G.H. Myotonic dystrophies type 1 and 2: A summary on current aspects. Semin. Pediatr. Neurol. 2006, 13, 71–79. [Google Scholar] [CrossRef]
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Santoro, M.; Fontana, L.; Masciullo, M.; Bianchi, M.L.E.; Rossi, S.; Leoncini, E.; Novelli, G.; Botta, A.; Silvestri, G. Expansion size and presence of CCG/CTC/CGG sequence interruptions in the expanded CTG array are independently associated to hypermethylation at the DMPK locus in myotonic dystrophy type 1 (DM1). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Ergoli, M.; Venditti, M.; Picillo, E.; Minucci, S.; Politano, L. Study of expression of genes potentially responsible for reduced fitness in patients with myotonic dystrophy type 1 and identification of new biomarkers of testicular function. Mol. Reprod. Dev. 2020, 87, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Charlet-B, N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019, 28, 1312–1321. [Google Scholar] [CrossRef]
- Anvret, M.; Ahlberg, G.; Grandell, U.; Hedberg, B.; Johnson, K.; Edstrom, L. Larger expansions of the CTG repeat in muscle compared to lymphocytes from patients with myotonic dystrophy. Hum. Mol. Genet. 1993, 2, 1397–1400. [Google Scholar] [CrossRef]
- Ashizawa, T.; Dunne, C.J.; Dubel, J.R.; Perryman, M.B.; Epstein, H.F.; Boerwinkle, E.; Hejtmancik, J.F. Anticipation in myotonic dystrophy: I. statistical verification based on clinical and haplotype findings. Neurology 1992, 42, 1871. [Google Scholar] [PubMed]
- Harley, H.G.; Rundle, S.A.; MacMillan, J.C.; Myring, J.; Brook, J.D.; Crow, S.; Reardon, W.; Fenton, I.; Shaw, D.J.; Harper, P.S. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum. Genet. 1993, 52, 1164. [Google Scholar]
- Cumming, S.A.; Jimenez-Moreno, C.; Okkersen, K.; Wenninger, S.; Daidj, F.; Hogarth, F.; Littleford, R.; Gorman, G.; Bassez, G.; Schoser, B.; et al. Genetic determinants of disease severity in the myotonic dystrophy type 1 OPTIMISTIC cohort. Neurology 2019, 93, e995–e1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G. Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry-based study with implications for disease classification. Rev. Neurol. 2016, 172, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; De Braekeleer, M.; Prévost, C.; Boily, C. Myotonic dystrophy: Clinical assessment of muscular disability in an isolated population with presumed homogeneous mutation. Neurology 1992, 42, 203. [Google Scholar] [CrossRef] [PubMed]
- Ballester-Lopez, A.; Koehorst, E.; Almendrote, M.; Martínez-Piñeiro, A.; Lucente, G.; Linares-Pardo, I.; Núñez-Manchón, J.; Guanyabens, N.; Cano, A.; Lucia, A.; et al. A DM1 family with interruptions associated with atypical symptoms and late onset but not with a milder phenotype. Hum. Mutat. 2020, 41, 420–431. [Google Scholar] [CrossRef]
- Pešović, J.; Perić, S.; Brkušanin, M.; Brajušković, G.; Rakočević-Stojanović, V.; Savić-Pavićević, D. Repeat interruptions modify age at onset in myotonic dystrophy type 1 by stabilizing DMPK expansions in somatic cells. Front. Genet. 2018, 9, 601. [Google Scholar] [CrossRef]
- Cumming, S.A.; Hamilton, M.J.; Robb, Y.; Gregory, H.; McWilliam, C.; Cooper, A.; Adam, B.; McGhie, J.; Hamilton, G.; Herzyk, P.; et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur. J. Hum. Genet. 2018, 26, 1635–1647. [Google Scholar] [CrossRef]
- Barbé, L.; Lanni, S.; López-Castel, A.; Franck, S.; Spits, C.; Keymolen, K.; Seneca, S.; Tomé, S.; Miron, I.; Letourneau, J.; et al. CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am. J. Hum. Genet. 2017, 100, 488–505. [Google Scholar]
- Légaré, C.; Overend, G.; Guay, S.-P.; Monckton, D.G.; Mathieu, J.; Gagnon, C.; Bouchard, L. DMPK gene DNA methylation levels are associated with muscular and respiratory profiles in DM1. Neurol. Genet. 2019, 5, e338. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, P.; Gläser, D.; Vogel, W.; Wolf, M.; Schwemmle, S. The DMPK gene of severely affected myotonic dystrophy patients is hypermethylated proximal to the largely expanded CTG repeat. Am. J. Hum. Genet. 1998, 62, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Spits, C.; Seneca, S.; Hilven, P.; Liebaers, I.; Sermon, K. Methylation of the CpG sites in the myotonic dystrophy locus does not correlate with CTG expansion size or with the congenital form of the disease. J. Med. Genet. 2010, 47, 700–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, A.L.; Nakamori, M.; Tomé, S.; Chitayat, D.; Gourdon, G.; Thornton, C.A.; Pearson, C.E. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 2011, 20, 1–15. [Google Scholar]
- Pešović, J.; Perić, S.; Brkušanin, M.; Brajušković, G.; Rakočević-Stojanović, V.; Savić-Pavićević, D. Molecular genetic and clinical characterization of myotonic dystrophy type 1 patients carrying variant repeats within DMPK expansions. Neurogenetics 2017, 18, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Masciullo, M.; Pietrobono, R.; Conte, G.; Modoni, A.; Bianchi, M.L.E.; Rizzo, V.; Pomponi, M.G.; Tasca, G.; Neri, G.; et al. Molecular, clinical, and muscle studies in myotonic dystrophy type 1 (DM1) associated with novel variant CCG expansions. J. Neurol. 2013, 260, 1245–1257. [Google Scholar] [PubMed]
- Musova, Z.; Mazanec, R.; Krepelova, A.; Ehler, E.; Vales, J.; Jaklova, R.; Prochazka, T.; Koukal, P.; Marikova, T.; Kraus, J.; et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. Part A 2009, 149, 1365–1374. [Google Scholar] [CrossRef]
- Tomé, S.; Dandelot, E.; Dogan, C.; Bertrand, A.; Geneviève, D.; Péréon, Y.; Simon, M.; Bonnefont, J.P.; Bassez, G.; Gourdon, G.; et al. Unusual association of a unique CAG interruption in 5′ of DM1 CTG repeats with intergenerational contractions and low somatic mosaicism. Hum. Mutat. 2018, 39, 970–982. [Google Scholar] [CrossRef]
- Braida, C.; Stefanatos, R.K.A.; Adam, B.; Mahajan, N.; Smeets, H.J.M.; Niel, F.; Goizet, C.; Arveiler, B.; Koenig, M.; Lagier-Tourenne, C.; et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2010, 19, 1399–1412. [Google Scholar]
- RStudio: Integrated Development for R; Rstudio, PBC: Boston, MA, USA, 2020.
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Shaw, D.J.; Chaudhary, S.; Rundle, S.A.; Crow, S.; Brook, J.D.; Harper, P.S.; Harley, H.G. A study of DNA methylation in myotonic dystrophy. J. Med. Genet. 1993, 30, 189–192. [Google Scholar]
- Morales, F.; Vásquez, M.; Corrales, E.; Vindas-Smith, R.; Santamaría-Ulloa, C.; Zhang, B.; Sirito, M.; Estecio, M.R.; Krahe, R.; Monckton, D.G. Longitudinal increases in somatic mosaicism of the expanded CTG repeat in myotonic dystrophy type 1 are associated with variation in age-at-onset. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef]
- Thornton, C.A.; Johnson, K.; Moxley, R.T. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol. 1994, 35, 104–107. [Google Scholar] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hildonen, M.; Knak, K.L.; Dunø, M.; Vissing, J.; Tümer, Z. Stable Longitudinal Methylation Levels at the CpG Sites Flanking the CTG Repeat of DMPK in Patients with Myotonic Dystrophy Type 1. Genes 2020, 11, 936. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080936
Hildonen M, Knak KL, Dunø M, Vissing J, Tümer Z. Stable Longitudinal Methylation Levels at the CpG Sites Flanking the CTG Repeat of DMPK in Patients with Myotonic Dystrophy Type 1. Genes. 2020; 11(8):936. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080936
Chicago/Turabian StyleHildonen, Mathis, Kirsten Lykke Knak, Morten Dunø, John Vissing, and Zeynep Tümer. 2020. "Stable Longitudinal Methylation Levels at the CpG Sites Flanking the CTG Repeat of DMPK in Patients with Myotonic Dystrophy Type 1" Genes 11, no. 8: 936. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080936