Combined PTPN11 and MYBPC3 Gene Mutations in an Adult Patient with Noonan Syndrome and Hypertrophic Cardiomyopathy

 ,
,
Abstract
:1. Background
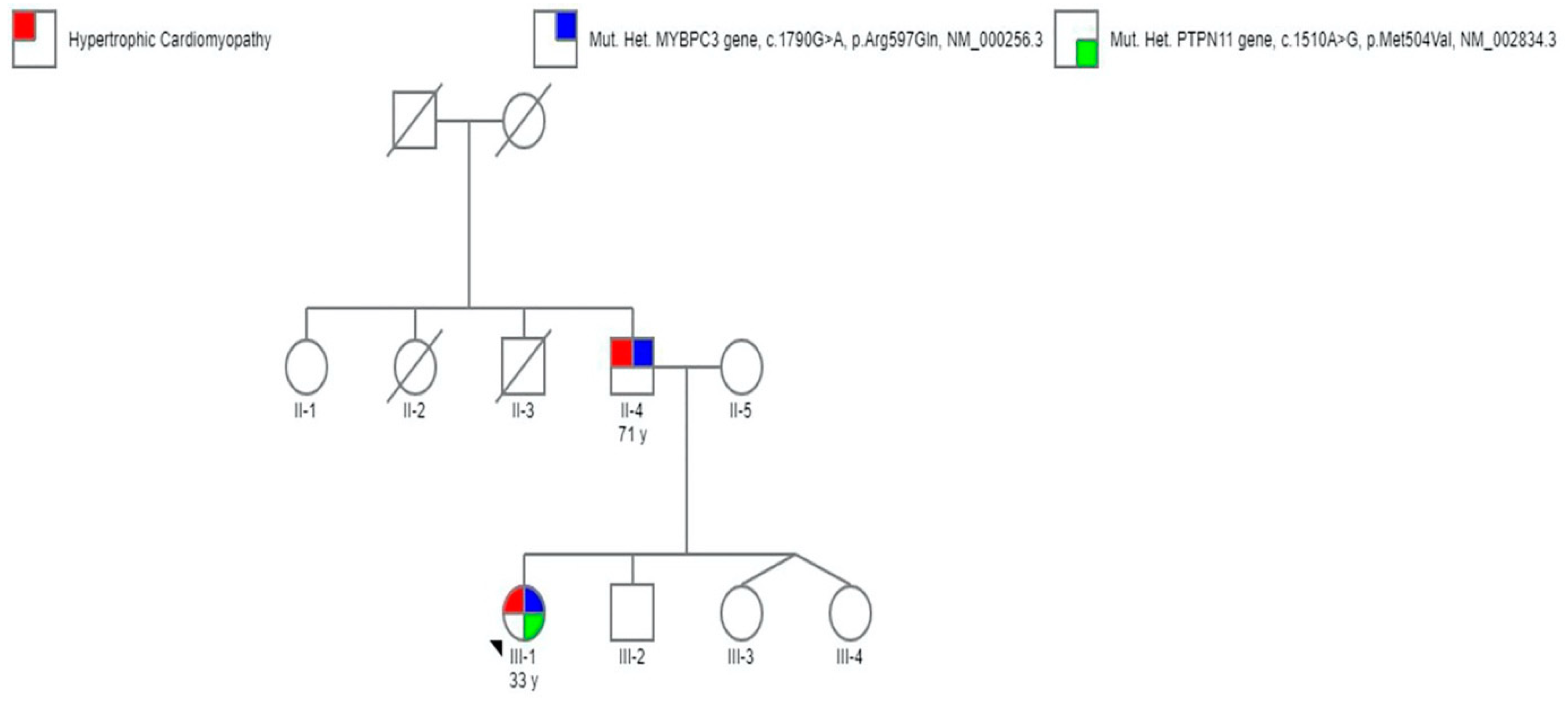
2. Case Report
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Esposito, A.; Monda, E.; Gragnano, F.; Simone, F.; Cesaro, A.; Natale, F.; Concilio, C.; Moscarella, E.; Caiazza, M.; Pazzanese, V.; et al. Prevalence and clinical implications of hyperhomocysteinaemia in patients with hypertrophic cardiomyopathy and MTHFR C6777T polymorphism. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Augusto, E.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Roberts, E.A.; Tartaglia, M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog. Pediatric Cardiol. 2015, 39, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, E.A.; Allanson, E.J.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Bouchikhi, I.; Belhassan, K.; Moufid, F.Z.; IraquiHoussaini, M.; Bouguenouch, L.; Samri, I.; Samir, A.; Ouldim, K. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int. J. Pediatrics Adolesc. Med. 2016, 3, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Kalidas, K.; Shaw, A.; Song, X.; Musat, D.L.; van der Burgt, I.; Brunner, H.; Bertola, D.R.; Crosby, A.; Ion, A.; et al. PTPN11 mutations in Noonan syndrome: Molecular spectrum genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 2002, 70, 1555–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncini, S.; Bonati, M.T.; Morella, I.; Ferrari, L.; Brambilla, R.; Riva, P. Differential allelic expression of SOS1 and hyperexpression of the activating SOS1 c.755c variant in a Noonan syndrome family. Eur. J. Hum. Genet. 2015, 23, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Trier, D.; Rinne, T.; Noordam, K.; Draaisma, J.M.; van der Burgt, I. Variable phenotypic expression in a large Noonan syndrome family segregating a novel SOS1 mutation. Am. J. Hum. Genet. 2017, 173, 2968–2972. [Google Scholar]
- Tekendo-Ngongang, C.; Agenbag, G.; DomilongoBope, C.; Esterhuizen, A.I.; Wonkam, A. Noonan syndrome in South Africa: Clinical and molecular profiles. Front. Genet. 2019, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Lafont, E.; Nony, S.; Rouvet, I.; Bozon, D. Functional characterization of putative novel splicing mutations in the cardiomyopathy-causing genes. DNA Cell Biol. 2015, 34, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.P.; Gómez-Molina, G.; Carreto-Alba, P.; Granell-Escobar, R.; Vázquez-Rico, I.; León-Justel, A. Noonan syndrome: Severe phenotype and PTPN11 mutations. Med. Clínica 2019, 152, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Girolami, F.; Ho, C.Y.; Semsarian, C.; Baldi, M.; Will, M.L.; Baldini, K.; Olivotto, I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010, 55, 1444–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cao, H.; Song, Y.; Feng, Y.; Ding, X.; Pang, M.; Zhang, Y.; Zhang, H.; Ding, J.; Xia, X. Identification of novel mutations including a double mutation in patients with inherited cardiomyopathy by a targeted sequencing approach using the Ion Torrent PGM system. Int. J. Mol. Med. 2016, 37, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alström syndrome. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; DArgenio, V.; Esposito, M.V.; Monda, E.; Maggio, F.D.; Frisso, G.; Salvatore, F. Genotype-phenotype correlation: A triple DNA mutational event in a boy entering sport conveys an additional pathogenicity risk. Genes 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, G.; Limongelli, G.; D’Ambrosio, A.; Gesualdo, F.; Digilio, M.C.; Baban, A.; Marino, B. Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int. J. Cardiol. 2017, 245, 92–98. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ID | Cardiological Features | Genetic Variants | ACMG Score | Gnomad Frequencies | ClinVar Classification |
|---|---|---|---|---|---|
| III.1 | HCM | PTPN11 gene (c.1510A>G, p.Met504Val, NM_002834.3) | PS3 | 0.00000882 | Pathogenic |
| MYBPC3 gene (c.1790G>A, p.Arg597Gln, NM_000256.3) | PM1 | 0.00000886 | Likely pathogenic | ||
| II.4 | HCM | PTPN11 WT/WT MYBPC3 gene (c.1790G>A, p.Arg597Gln, NM_000256.3) | PM1 | 0.00000886 | Pathogenic |
| II.5 | No evidence of cardiovascular abnormalities | PTPN11 WT/WT MYBPC3 WT/WT | N/A | N/A | N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caiazza, M.; Rubino, M.; Monda, E.; Passariello, A.; Fusco, A.; Cirillo, A.; Esposito, A.; Pierno, A.; De Fazio, F.; Pacileo, R.; et al. Combined PTPN11 and MYBPC3 Gene Mutations in an Adult Patient with Noonan Syndrome and Hypertrophic Cardiomyopathy. Genes 2020, 11, 947. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080947
Caiazza M, Rubino M, Monda E, Passariello A, Fusco A, Cirillo A, Esposito A, Pierno A, De Fazio F, Pacileo R, et al. Combined PTPN11 and MYBPC3 Gene Mutations in an Adult Patient with Noonan Syndrome and Hypertrophic Cardiomyopathy. Genes. 2020; 11(8):947. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080947
Chicago/Turabian StyleCaiazza, Martina, Marta Rubino, Emanuele Monda, Annalisa Passariello, Adelaide Fusco, Annapaola Cirillo, Augusto Esposito, Anna Pierno, Federica De Fazio, Roberta Pacileo, and et al. 2020. "Combined PTPN11 and MYBPC3 Gene Mutations in an Adult Patient with Noonan Syndrome and Hypertrophic Cardiomyopathy" Genes 11, no. 8: 947. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080947