Comparison of Illumina versus Nanopore 16S rRNA Gene Sequencing of the Human Nasal Microbiota

 , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Collection and Selection
2.2. DNA Isolation
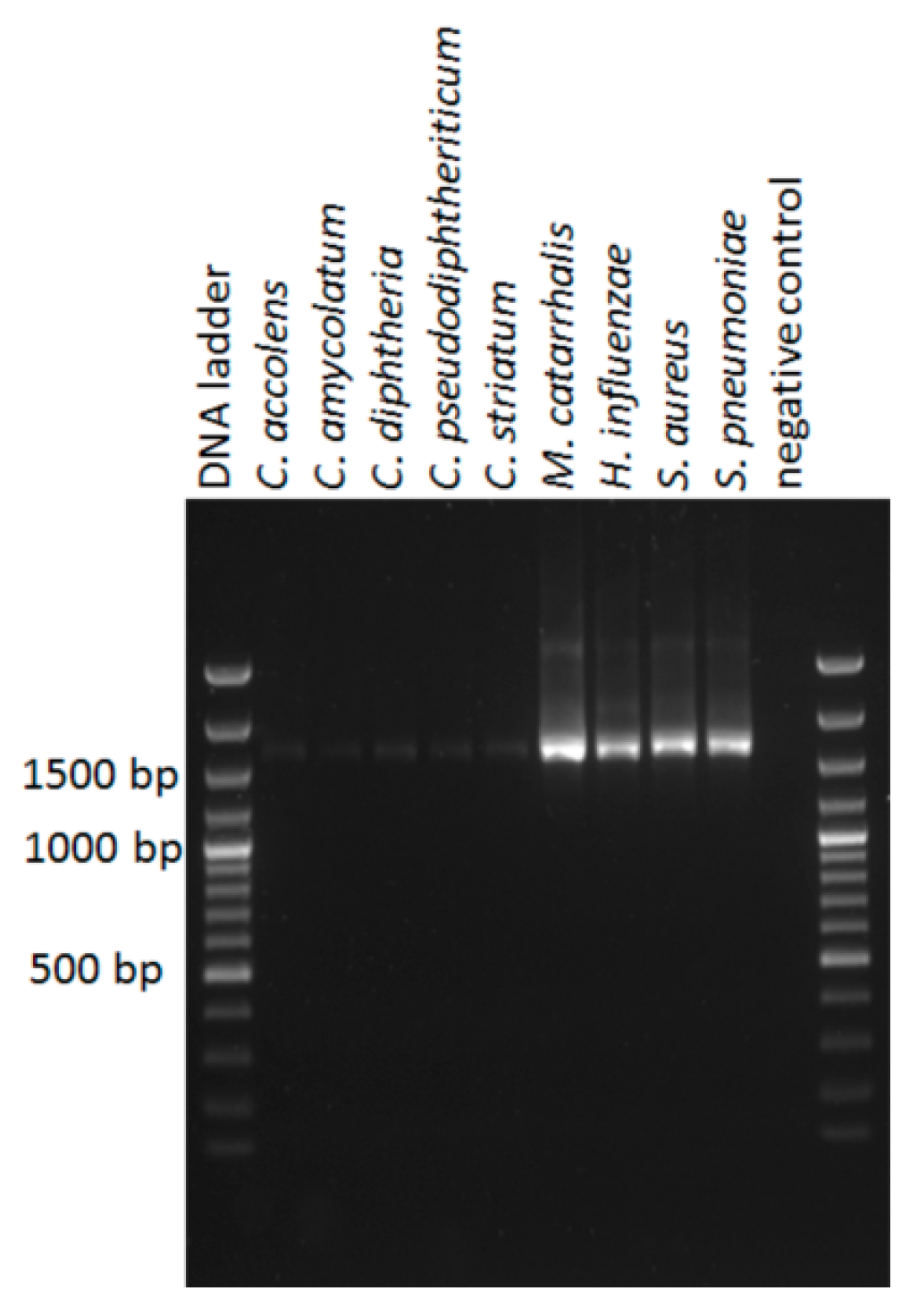
2.3. Bacterial Strains
2.4. Illumina Sequencing
2.5. Nanopore Sequencing
2.6. Data Analysis
2.7. Statistics
2.8. Sequence Data Availability
3. Results
3.1. Sample Population
3.2. General Sequencing Results
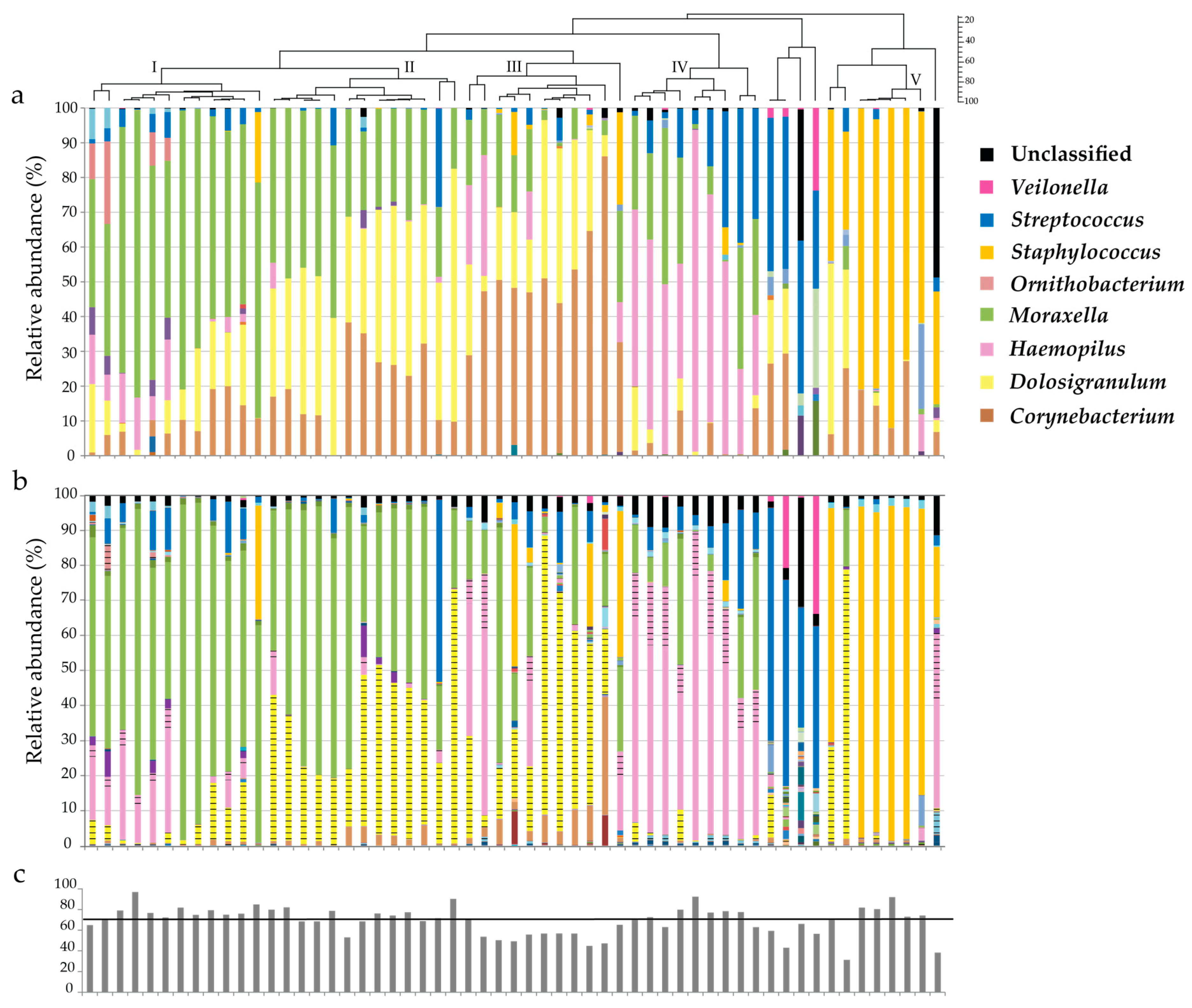
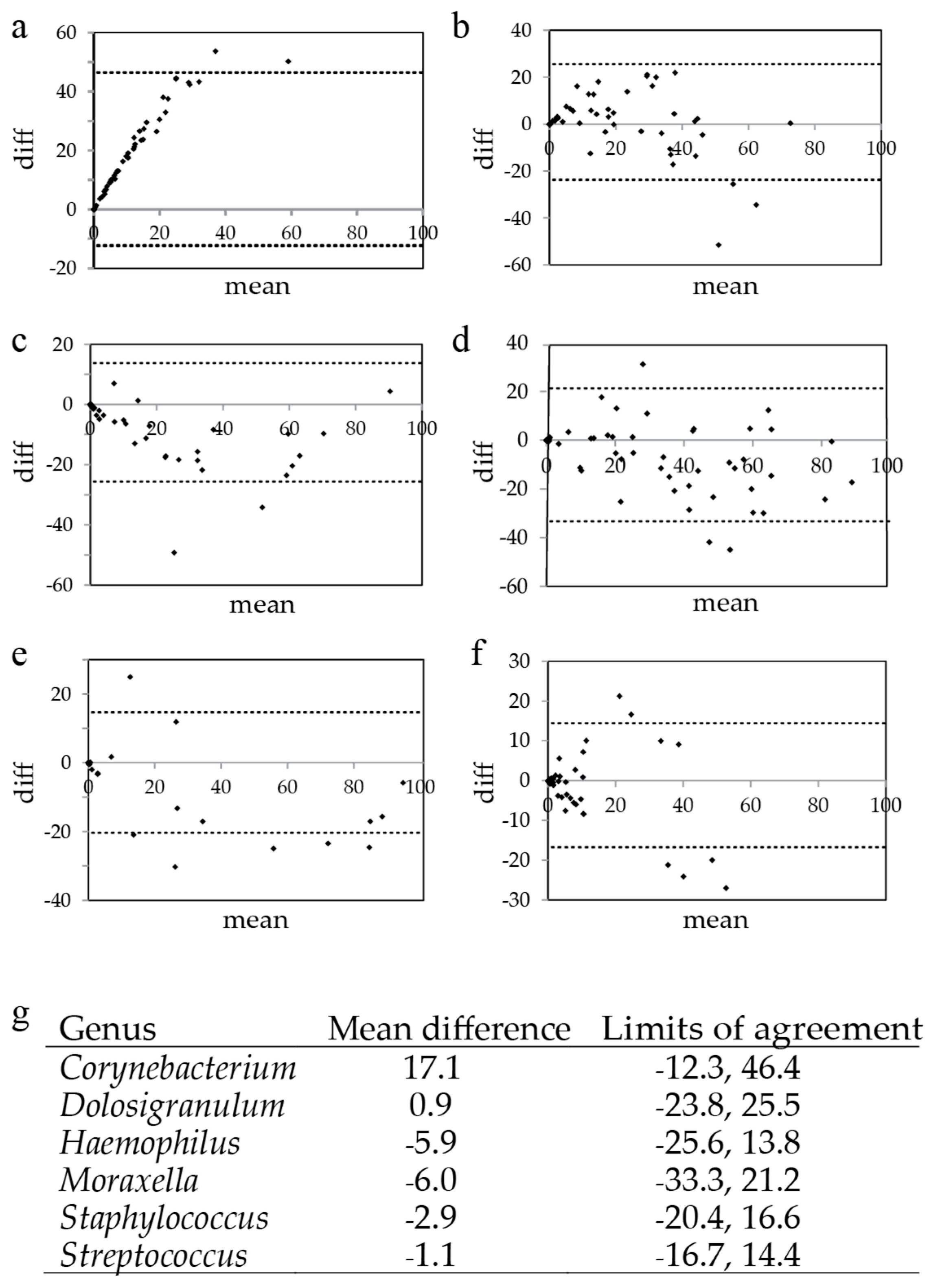
3.3. Illumina versus Nanopore Sequencing
3.4. Prevalence of Corynebacterium spp.
3.5. Re-Basecalling and Analysis of the Nose Swab Samples
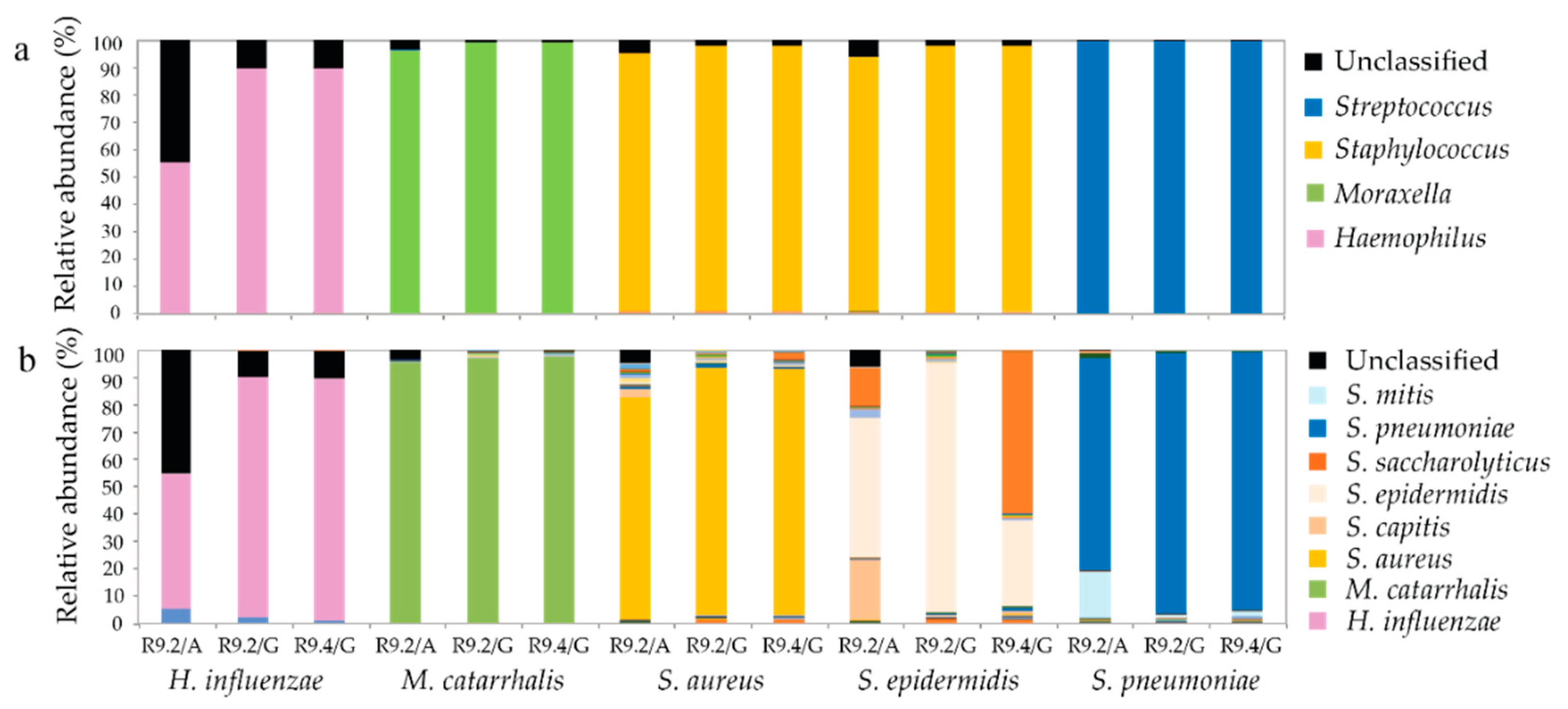
3.6. Genus and Species Level Taxonomy on Pure Cultured Single Species Bacteria Using Nanopore Sequencing
4. Discussion
5. Conclusions
Ethics Approval and Consent to Participate
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mansbach, J.M.; Luna, P.N.; Shaw, C.A.; Hasegawa, K.; Petrosino, J.F.; Piedra, P.A.; Sullivan, A.F.; Espinola, J.A.; Stewart, C.J.; Camargo, C.A., Jr. Increased Moraxella and Streptococcus species abundance after severe bronchiolitis is associated with recurrent wheezing. J. Allergy Clin. Immunol. 2020, 145, 518–527.e8. [Google Scholar] [CrossRef] [Green Version]
- Bomar, L.; Brugger, S.D.; Lemon, K.P. Bacterial microbiota of the nasal passages across the span of human life. Curr. Opin. Microbiol. 2017, 41, 8–14. [Google Scholar] [CrossRef]
- Mika, M.; Korten, I.; Qi, W.; Regamey, N.; Frey, U.; Casaulta, C.; Latzin, P.; Hilty, M.; SCILD Study Group. The nasal microbiota in infants with cystic fibrosis in the first year of life: A prospective cohort study. Lancet Respir. Med. 2016, 4, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Hui, J.W.; Ong, J.; Herdegen, J.J.; Kim, H.; Codispoti, C.D.; Kalantari, V.; Tobin, M.C.; Schleimer, R.P.; Batra, P.S.; LoSavio, P.S.; et al. Risk of obstructive sleep apnea in African American patients with chronic rhinosinusitis. Ann. Allergy Asthma Immunol. 2017, 118, 685–688.e1. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Ajami, N.J.; Ghantoji, S.S.; Shelburne, S.; El_Haddad, D.; Shah, P.; Piedra, P.; Shpall, E.; Kontoyiannis, D.P.; Chemaly, R.F. Nasal Microbiota Changes are Associated with Progression to Lower Respiratory Infection Following Respiratory Syncytial Virus Upper Respiratory Infection in Hematopoietic Cell Transplant Recipients. Open Forum Infect. Dis. 2016, 3, 2232. [Google Scholar] [CrossRef] [Green Version]
- Man, W.H.; de Steenhuijsen Piters, W.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Man, W.H.; van Houten, M.A.; Mérelle, M.E.; Vlieger, A.M.; Chu, M.L.J.N.; Jansen, N.J.G.; Sanders, E.A.M.; Bogaert, D. Bacterial and viral respiratory tract microbiota and host characteristics in children with lower respiratory tract infections: A matched case-control study. Lancet Respir. Med. 2019, 7, 417–426. [Google Scholar] [CrossRef]
- Lu, Y.J.; Sasaki, T.; Kuwahara-Arai, K.; Uehara, Y.; Hiramatsu, K. Development of new application for comprehensive viability analysis based on microbiome analysis by next-generation sequencing: Insights into staphylococcal carriage in human nasal cavities. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Sadowsky, M.J.; Staley, C.; Heiner, C.; Hall, R.; Kelly, C.R.; Brandt, L.; Khoruts, A. Analysis of gut microbiota—An ever changing landscape. Gut Microbes 2017, 8, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Rohde, H.; Burandt, E.C.; Siemssen, N.; Frommelt, L.; Burdelski, C.; Wurster, S.; Scherpe, S.; Davies, A.P.; Harris, L.G.; Horstkotte, M.A.; et al. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials 2007, 28, 1711–1720. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Available online: https://nanoporetech.com/ (accessed on 1 June 2020).
- Shin, J.; Lee, S.; Go, M.J.; Lee, S.Y.; Kim, S.C.; Lee, C.H.; Cho, B.K. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci. Rep. 2016, 6, 29681. [Google Scholar] [CrossRef] [Green Version]
- Cusco, A.; Vines, J.; D’Andreano, S.; Riva, F.; Casellas, J.; Sanchez, A.; Francino, O. Using MinION™ to characterize dog skin microbiota through full-length 16S rRNA gene sequencing approach. bioRxiv 2017, 167015. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, S.; Kryukov, K.; Nakagawa, S.; Takeuchi, J.S.; Shiraishi, Y.; Asano, K.; Imanishi, T. A portable system for rapid bacterial composition analysis using a nanopore-based sequencer and laptop computer. Sci. Rep. 2017, 7, 5657. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, C.B.; Oved, K.; Eden, E.; Cohen, A.; Engelhard, D.; Boers, S.; Kraaij, R.; Karlsson, R.; Fernandez, D.; Gonzalez, E.; et al. Observational multi-centre, prospective study to characterize novel pathogen-and host-related factors in hospitalized patients with lower respiratory tract infections and/or sepsis-the “TAILORED-Treatment” study. BMC Infect. Dis. 2018, 18, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikema, A.; de Koning, W.; Li, Y.; Stubbs, A.; Hays, J.P. Lessons learnt from the introduction of nanopore sequencing? Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef]
- Yang, S.; Lin, S.; Kelen, G.D.; Quinn, T.C.; Dick, J.D.; Gaydos, C.A.; Rothman, R.E. Quantitative multiprobe PCR assay for simultaneous detection and identification to species level of bacterial pathogens. J. Clin. Microbiol. 2002, 40, 3449–3454. [Google Scholar] [CrossRef] [Green Version]
- Bogaert, D.; Keijser, B.; Huse, S.; Rossen, J.; Veenhoven, R.; van Gils, E.; Bruin, J.; Montijn, R.; Bonten, M.; Sanders, E. Variability and diversity of nasopharyngeal microbiota in children: A metagenomic analysis. PLoS ONE 2011, 6, e17035. [Google Scholar] [CrossRef] [Green Version]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batut, B.; Gravouil, K.; Defois, C.; Hiltemann, S.; Brugere, J.F.; Peyretaillade, E.; Peyret, P. ASaiM: A Galaxy-based framework to analyze microbiota data. Gigascience 2018, 7, giy057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva Reference Files. Available online: https://www.Mothur.org/wiki/Silva_reference_files (accessed on 29 August 2020).
- Oxford Nanopore Technologies EPI2ME. Available online: https://epi2me.nanoporetech.com/ (accessed on 29 August 2020).
- NCBI BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=TargLociBlast (accessed on 29 August 2020).
- Python Library ete2. Available online: https://pypi.org/project/ete2/ (accessed on 29 August 2020).
- European Nucleotide Archive (ENA). Available online: https://www.ebi.ac.uk/ena/data/search?query=PRJEB28612 (accessed on 11 September 2020).
- De Boeck, I.; Wittouck, S.; Wuyts, S.; Oerlemans, E.F.M.; van den Broek, M.F.L.; Vandenheuvel, D.; Vanderveken, O.; Lebeer, S. Comparing the Healthy Nose and Nasopharynx Microbiota Reveals Continuity As Well As Niche-Specificity. Front. Microbiol. 2017, 8, 2372. [Google Scholar] [CrossRef] [Green Version]
- Brugger, S.D.; Bomar, L.; Lemon, K.P. Commensal-Pathogen Interactions along the Human Nasal Passages. PLoS Pathog. 2016, 12, e1005633. [Google Scholar] [CrossRef] [Green Version]
- Biswas, K.; Hoggard, M.; Jain, R.; Taylor, M.W.; Douglas, R.G. The nasal microbiota in health and disease: Variation within and between subjects. Front. Microbiol. 2015, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Kai, S.; Matsuo, Y.; Nakagawa, S.; Kryukov, K.; Matsukawa, S.; Tanaka, H.; Iwai, T.; Imanishi, T.; Hirota, K. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinION nanopore sequencer. FEBS Open Bio 2019, 9, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Laursen, M.F.; Dalgaard, M.D.; Bahl, M.I. Genomic GC-Content Affects the Accuracy of 16S rRNA Gene Sequencing Based Microbial Profiling due to PCR Bias. Front. Microbiol. 2017, 8, 1934. [Google Scholar] [CrossRef] [Green Version]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [Google Scholar] [CrossRef]
- Bessesen, M.T.; Kotter, C.V.; Wagner, B.D.; Adams, J.C.; Kingery, S.; Benoit, J.B.; Robertson, C.E.; Janoff, E.N.; Frank, D.N. MRSA colonization and the nasal microbiome in adults at high risk of colonization and infection. J. Infect. 2015, 71, 649–657. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Information | Illumina Technology | Nanopore Technology | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Infection | Age (Years) | 16S Copies | Raw Reads | Percentage Reads Classified (%) (a) | Genera Identified ≥1% | Genera (ISI) | Raw Reads | Percentage Reads Classified (%) | Genera Identified ≥1% | Genera (ISI) |
| 1 | yes | 3.50 | 5 × 105 | 133,880 | 92 | 5 | 4.2 | 34,944 | 77 | 5 | 3.0 |
| 2 | yes | 0.92 | 1 × 105 | 186,250 | 95 | 5 | 1.9 | 15,254 | 79 | 3 | 2.2 |
| 3 | yes | 2.00 | 3 × 105 | 1661 | 94 | 5 | 4.1 | 39,474 | 77 | 4 | 2.9 |
| 4 | yes | 1.50 | 3 × 105 | 154,877 | 96 | 7 | 4.6 | 36,608 | 76 | 6 | 2.3 |
| 5 | yes | 9.00 | 3 × 105 | 114,702 | 97 | 5 | 3.5 | 5107 | 59 | 4 | 2.7 |
| 6 | yes | 2.00 | 3 × 105 | 22,805 | 97 | 5 | 2.7 | 31,642 | 52 | 4 | 1.7 |
| 7 | yes | 5.00 | 2 × 105 | 1940 | 88 | 8 | 3.8 | 2246 | 57 | 6 | 3.1 |
| 8 | yes | 4.00 | 3 × 105 | 24,214 | 100 | 4 | 1.2 | 10,174 | 62 | 3 | 1.2 |
| 9 | yes | 1.67 | 4 × 105 | 104,134 | 93 | 9 | 2.5 | 21,462 | 68 | 6 | 2.6 |
| 10 | yes | 8.00 | 2 × 105 | 186,945 | 96 | 3 | 2.5 | 923 | 68 | 2 | 1.6 |
| 11 | yes | 11.00 | 2 × 105 | 120,867 | 95 | 3 | 3.0 | 27,569 | 78 | 3 | 1.6 |
| 12 | yes | 0.42 | 4 × 105 | 25,743 | 98 | 3 | 3.0 | 5127 | 66 | 3 | 2.2 |
| 13 | yes | 15.00 | 4 × 105 | 261,123 | 95 | 4 | 2.7 | 12,572 | 66 | 5 | 2.0 |
| 14 | yes | 2.17 | 1 × 105 | 6246 | 97 | 4 | 3.0 | 20,441 | 89 | 3 | 2.7 |
| 15 | yes | 3.80 | 3 × 105 | 68,095 | 91 | 3 | 2.3 | 27,077 | 90 | 4 | 2.5 |
| 16 | yes | 2.40 | 1 × 105 | 119,295 | 84 | 7 | 2.9 | 2978 | 85 | 6 | 2.6 |
| 17 | yes | 0.80 | 2 × 105 | 74,902 | 96 | 3 | 1.5 | 4408 | 91 | 2 | 1.1 |
| 18 | yes | 61.00 | 3 × 103 | 77,851 | 86 | 6 | 3.4 | 2141 | 82 | 8 | 4.1 |
| 19 | yes | 0.90 | 3 × 105 | 74,730 | 85 | 4 | 2.3 | 20,584 | 82 | 6 | 1.6 |
| 20 | yes | 0.80 | 3 × 105 | 113,078 | 93 | 3 | 2.4 | 10,974 | 91 | 3 | 1.9 |
| 21 | yes | 78.00 | 2 × 105 | 131,837 | 90 | 2 | 1.7 | 21,449 | 93 | 1 | 1.0 |
| 22 | yes | 1.70 | 3 × 105 | 162,890 | 85 | 4 | 2.4 | 23,530 | 92 | 5 | 1.8 |
| 23 | yes | 2.30 | 2 × 105 | 83,596 | 92 | 8 | 4.4 | 15,748 | 88 | 7 | 3.2 |
| 24 | yes | 73.00 | 2 × 105 | 83,947 | 84 | 4 | 2.0 | 3181 | 88 | 5 | 3.3 |
| 25 | yes | 2.60 | 5 × 105 | 28,221 | 92 | 3 | 3.0 | 15,453 | 50 | 3 | 3.1 |
| 26 | yes | 65.00 | 3 × 105 | 77,012 | 82 | 7 | 4.5 | 31,461 | 85 | 6 | 2.8 |
| 27 | yes | 0.80 | 1 × 105 | 58,962 | 85 | 3 | 2.5 | 23,652 | 90 | 3 | 1.5 |
| 28 | yes | 3.00 | 5 × 105 | 57,600 | 86 | 6 | 3.7 | 22,991 | 84 | 7 | 3.4 |
| 29 | yes | 57.00 | 2 × 105 | 129,131 | 94 | 2 | 1.5 | 48,167 | 90 | 1 | 1.1 |
| 30 | yes | 0.40 | 6 × 105 | 180,796 | 88 | 3 | 2.9 | 3997 | 65 | 4 | 2.3 |
| 31 | yes | 0.90 | 4 × 105 | 547,695 | 98 | 4 | 2.7 | 15,626 | 80 | 7 | 1.7 |
| 32 | yes | 23.00 | 8 × 105 | 750,669 | 97 | 3 | 1.8 | 6653 | 67 | 2 | 1.6 |
| 33 | yes | 3.40 | 1 × 105 | 924,890 | 98 | 7 | 3.3 | 25,148 | 74 | 7 | 2.1 |
| 34 | yes | 4.10 | 1 × 106 | 31,896 | 94 | 5 | 4.0 | 15,979 | 49 | 4 | 2.7 |
| 35 | yes | 14.00 | 3 × 105 | 79,970 | 90 | 3 | 2.1 | 40,551 | 88 | 3 | 1.4 |
| 36 | yes | 0.10 | 3 × 105 | 113,047 | 88 | 3 | 1.7 | 50 | 76 | NA | NA |
| 37 (b) | yes | 0.40 | 3 × 105 | 59,397 | 88 | 4 | 2.9 | 51,254 | 63 | 11 | 3.6 |
| 38 | yes | 0.30 | 6 × 105 | 7421 | 99 | 3 | 1.4 | 41,757 | 89 | 2 | 1.4 |
| 39 | yes | 1.10 | 3 × 105 | 121,819 | 86 | 3 | 2.6 | 6340 | 86 | 6 | 1.9 |
| 40 | yes | 0.20 | 2 × 105 | 83,457 | 83 | 4 | 2.4 | 59,923 | 82 | 6 | 1.9 |
| 41 | yes | 4.20 | 4 × 105 | 92,006 | 87 | 4 | 2.9 | 17,785 | 90 | 4 | 2.3 |
| 42 | yes | 0.10 | 1 × 105 | 36,248 | 90 | 4 | 2.0 | 45,047 | 92 | 3 | 1.9 |
| 43 | yes | 0.10 | 2 × 105 | 55,585 | 92 | 5 | 2.3 | 47,084 | 92 | 3 | 1.4 |
| 44 | yes | 0.40 | 3 × 105 | 101,465 | 87 | 5 | 2.7 | 5288 | 80 | 6 | 1.6 |
| 45 | yes | 1.70 | 7 × 105 | 92,476 | 89 | 3 | 1.9 | 49,104 | 55 | 2 | 1.1 |
| 46 | yes | 0.50 | 3 × 105 | 72,068 | 88 | 4 | 2.1 | 50,486 | 80 | 6 | 1.5 |
| 47 | yes | 0.10 | 5 × 105 | 90,128 | 80 | 6 | 4.0 | 107,161 | 91 | 6 | 3.2 |
| 48 (b) | yes | 67.00 | 2 × 105 | 51,826 | 94 | 5 | 1.3 | 8 | 75 | NA | NA |
| 49 | yes | 0.30 | 9 × 105 | 1148 | 82 | 8 | 4.3 | 14,673 | 66 | 3 | 1.5 |
| 50 | yes | 3.30 | 5 × 106 | 39,030 | 83 | 3 | 2.6 | 12,239 | 66 | 3 | 2.0 |
| 51 | yes | 56.00 | 5 × 106 | 2191 | 85 | 7 | 3.3 | 17,248 | 64 | 7 | 2.4 |
| 52 | no | 28.00 | 3 × 105 | 193,859 | 96 | 2 | 1.2 | 6,789 | 91 | 1 | 1.0 |
| 53 | no | 62.00 | 2 × 105 | 262,184 | 89 | 3 | 2.3 | 18,680 | 88 | 2 | 1.8 |
| 54 | no | 8.10 | 2 × 105 | 308,123 | 83 | 5 | 2.8 | 13,741 | 89 | 4 | 2.3 |
| 55 | no | 7.20 | 3 × 105 | 203,242 | 100 | 6 | 3.4 | 15,490 | 84 | 6 | 4.3 |
| 56 | no | 14.90 | 1 × 105 | 235,820 | 92 | 3 | 1.4 | 18,318 | 88 | 8 | 5.0 |
| 57 | no | 5.40 | 9 × 105 | 90,422 | 86 | 3 | 2.8 | 11,207 | 87 | 4 | 2.2 |
| 58 | no | 7.10 | 6 × 105 | 103,176 | 87 | 5 | 2.9 | 19,604 | 73 | 7 | 2.7 |
| 59 | no | 6.40 | 1 × 105 | 111,844 | 93 | 4 | 1.6 | 17,971 | 88 | 3 | 1.1 |
| Average | NA | 12.5 | 8× 105 | 131,024 | 91 | 4.4 | 2.7 | 21,907 | 78 | 4.5 | 2.2 |
| Control | |||||||||||
| C-1 | NA | NA | <1 × 102 | 6 | 0 | 0 | NA | 7 | 57 | 4 | NA |
| C-2 | NA | NA | <1 × 102 | 1 | 0 | 0 | 0 | 42 | 74 | 8 | NA |
| C-3 | NA | NA | <1 × 102 | 1 | 0 | 0 | 0 | 33 | 42 | 9 | NA |
| C-4 | NA | NA | <1 × 102 | 3 | 0 | 0 | 0 | 35 | 51 | 11 | NA |
| C-5 | NA | NA | <1 × 102 | 2 | 0 | 0 | 0 | 15 | 67 | 3 | NA |
| C-6 | NA | NA | 2 × 102 | 2440 | 98 | 4 | 4 | 56 | 91 | 6 | NA |
| C-7 | NA | NA | 3 × 102 | 3408 | 94 | 18 | 18 | 0 | 0 | 0 | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heikema, A.P.; Horst-Kreft, D.; Boers, S.A.; Jansen, R.; Hiltemann, S.D.; de Koning, W.; Kraaij, R.; de Ridder, M.A.J.; van Houten, C.B.; Bont, L.J.; et al. Comparison of Illumina versus Nanopore 16S rRNA Gene Sequencing of the Human Nasal Microbiota. Genes 2020, 11, 1105. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091105
Heikema AP, Horst-Kreft D, Boers SA, Jansen R, Hiltemann SD, de Koning W, Kraaij R, de Ridder MAJ, van Houten CB, Bont LJ, et al. Comparison of Illumina versus Nanopore 16S rRNA Gene Sequencing of the Human Nasal Microbiota. Genes. 2020; 11(9):1105. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091105
Chicago/Turabian StyleHeikema, Astrid P., Deborah Horst-Kreft, Stefan A. Boers, Rick Jansen, Saskia D. Hiltemann, Willem de Koning, Robert Kraaij, Maria A. J. de Ridder, Chantal B. van Houten, Louis J. Bont, and et al. 2020. "Comparison of Illumina versus Nanopore 16S rRNA Gene Sequencing of the Human Nasal Microbiota" Genes 11, no. 9: 1105. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091105