Suprabasin—A Review


Laboratory of Genome Integrity, Institute of Molecular Genetics of the Czech Academy of Sciences, Videnska 1083, 14220 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Genes 2021, 12(1), 108; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010108
Submission received: 17 December 2020
/
Revised: 11 January 2021
/
Accepted: 13 January 2021
/
Published: 18 January 2021
(This article belongs to the Section Human Genomics and Genetic Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Among the ~22,000 human genes, very few remain that have unknown functions. One such example is suprabasin (SBSN). Originally described as a component of the cornified envelope, the function of stratified epithelia-expressed SBSN is unknown. Both the lack of knowledge about the gene role under physiological conditions and the emerging link of SBSN to various human diseases, including cancer, attract research interest. The association of SBSN expression with poor prognosis of patients suffering from oesophageal carcinoma, glioblastoma multiforme, and myelodysplastic syndromes suggests that SBSN may play a role in human tumourigenesis. Three SBSN isoforms code for the secreted proteins with putative function as signalling molecules, yet with poorly described effects. In this first review about SBSN, we summarised the current knowledge accumulated since its original description, and we discuss the potential mechanisms and roles of SBSN in both physiology and pathology.
1. Introduction
Since its original description in human and mouse keratinocyte differentiation [1], suprabasin (SBSN) has been associated with multiple diseases, including cancer. SBSN isoforms are putative signalling molecules inducing cellular signalling (AKT, WNT/β-catenin, and/or p38MAPK signalling) and various cellular processes, such as migration, proliferation, neovascularization, therapy-, apoptosis- and immune-resistance. Therefore, SBSN is considered an oncogene and is a proposed biomarker in a couple of diseases, lung carcinoma and myelodysplastic syndromes (MDS). The apparent need for a deeper understating of the nature and the function of SBSN prompted us to compile all current knowledge of SBSN together with suggestions for future research work.
2. SBSN Gene Organization
SBSN gene is located on human chromosome 19 (chr 19: 35,523,367–35,528,351 reverse strand; GRCh38:CM000681.2; band 19q13.1) close to other keratinocyte-differentiation associated genes dermokine-α/β and KDAP [1,2]. In mice it corresponds to chromosome 7 (7:30,751,471–30,756,134 forward strand; GRCm38:CM001000.2; band 7B2–7B3). The SBSN gene is a part of a coordinately expressed new stratified epithelium-related gene cluster, tentatively named stratified epithelium secreted peptides complex, SSC [2]. SBSN was studied mostly in human and mice; however, other mammals possess SBSN homologs. Predicted SBSN peptide encoded in the genome of Gorilla gorilla gorilla possesses 97.6% amino acid sequence similarity to the human SBSN-1. Relatively high amino acid sequence identity (58.9%) between mouse and human orthologs of the largest SBSN isoform (SBSN isoform 1; SBSN-1) suggests strong gene integrity and conserved function among the species. Paralogs of SBSN were not defined, and no genes with confidently high sequence identities were identified. The Ensembl database [3] (Ensembl Genome Browser version 101, accessed on 21 August 2020) enlists 106 SBSN orthologs (26/26 primates; 30/32 rodents and related; 39/45 Laurasiatheria; 0/19 Sauropsida; 2/86 fish, and nine Monotremata and Marsupialia; Figure 1) with 96 orthologs having Gene Order Conservation Score 100 (identical four closest genes), implicating true orthology. Note, the putative SBSN fish genes show Gene Order Conservation Scores of 0 and 2.1% and 8.6% sequence identity to the human ortholog, respectively. Altogether, these observations suggest that SBSN is likely a mammalian-specific orphan gene.
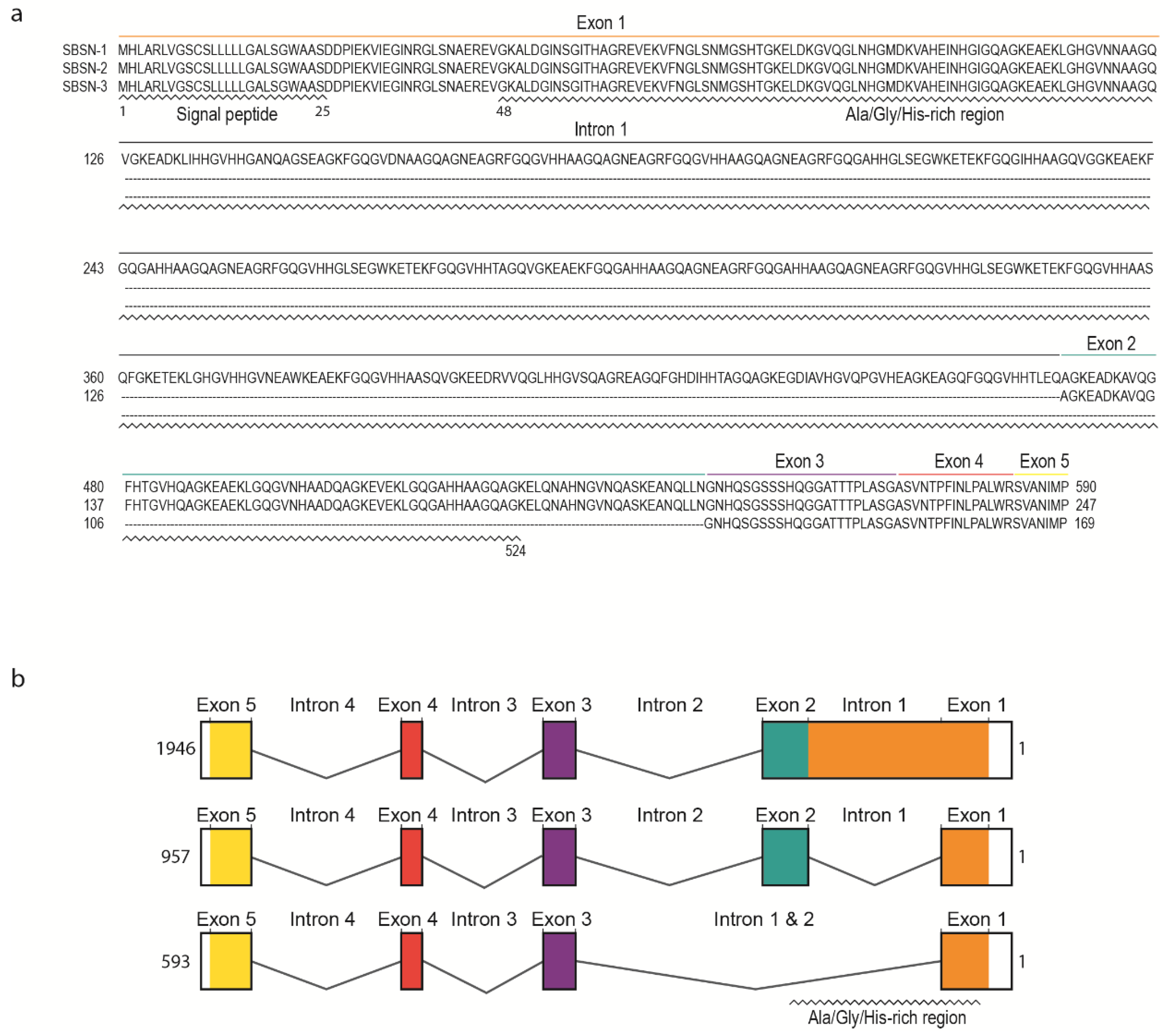
Human SBSN gene consists of five exons and four introns (Figure 2a). Human SBSN mRNA can be alternatively spliced producing three known isoforms SBSN-1 (transcript length: 1946 bp; ENST00000452271.7), SBSN-2 (957 bp; ENST00000518157.1) and SBSN-3 (593 bp; ENST00000588674.5). Importantly, SBSN-2 represents a fully spliced isoform [5], whereas SBSN-1 contains an in-frame retention of the first intron. SBSN-3 is also a fully spliced isoform but lacks exon 2. Strikingly, the mouse (Mus musculus) isoform corresponding to human SBSN-2 has not been identified. This may restrict the applicability of a mouse model for functional studies of SBSN isoforms. Interestingly, a sequence with high sequence identity (81.4%) to human exon two is present within the murine Sbsn-1 exon one, and the major difference between the human intron one exon two junction and homologous murine sequence (CACGAGGCCGGG vs. AACCAGGGTCAA) implies that in mice, the splicing site was likely not established, or was lost. The murine putative exon two is spliced together with the entire exon one in murine Sbsn-2, hence resembling human isoform three, based upon amino acids sequence identity analysis (44.5% for human SBSN-2 vs. murine Sbsn-2, and 65.5% for human SBSN-3 vs. murine Sbsn-2). Multiple other rodents lack homolog corresponding to the human SBSN-2 isoform, and interestingly, several primates (e.g., Macaca mulatta, Pan paniscus and Microcebus murinus) lack putative SBSN-2 as well. However, this may be due to the absence of its identification, rather than sequence deviation.
3. SBSN Protein Structure
The UniProtKB database [6] (The UniProt release 2020_4, accessed 21 August 2020) refers to three human SBSN isoforms (Figure 2b). SBSN-1 (Q6UWP8-1; 590 aa, predicted mass 60.541 Da) and SBSN-2 (Q6UWP8-2; 247 aa, predicted mass 25.335 Da) are well defined. However, SBSN-3 (K7ESC4) is described as a 149 aa long peptide with a predicted mass of 15.318 Da, which is incorrect due to the lacking description of putative N-terminal signal peptide (i.e., missing N-terminal sequence MHLARLVGSCSLLLLLGALS; see also sequence alignment in Figure 2a) in the database. In fact, the SBSN-3 coding sequence is 507 nts long and translates into a 169 aa long peptide. All SBSN isoforms possess a putative N-terminal signal peptide (aa 1–25) [2,7] granting SBSN secretory nature and extracellular localization.
Distinctly of the other two isoforms, SBSN-1 is alanine-(14%), glycine-(20%), and histidine-(10%) rich due to the presence of short tandem Glycine-x-Histidine-Histidine repeats (GxHH repeats, x stands for any classical amino acid) encoded by the retained intron one. Note, structural proteins often contain compositional biases of similar characteristics to SBSN-1 Ala-/Gly-/His-rich domain (aa 26—524), and based upon these observations, SBSN was proposed as a component of the cornified envelope (CE), the essential structure of corneocytes responsible for the skin protective function [1,7]. Notably, no similar sequence encoding SBSN-1 Ala-/Gly-/His-rich domain is present in the human genome, indicating an evolutionarily unique and conserved role. This is further supported by a comparison of SBSN among species, showing remarkable conservation of the Ala-/Gly-/His-rich domain, as well as retention of the first intron encoding the domain. Human SBSN-2 and SBSN-3 lack most of the Ala-/Gly-/His-rich domains due to intron one splicing; however, a short repeat is located at the 3′ end of exon one. Similarly to Ala-/Gly-/His-rich domain of SBSN-1, the C-terminal sequence of all isoforms is of unknown function. As mentioned, SBSN-3 lacks exon two, but the remaining exons are present in all human SBSN isoforms.
The mouse homolog of SBSN possesses similar features. The N-terminal signal peptide was predicted (aa 1–23), and the compositional bias of Ala-/Gly-/His-rich domain (aa 97—478) in SBSN-1 (Q8CIT9; 700 aa; predicted mass 72.334 Da) is also present in mouse homolog. The current description of other murine Sbsn isoforms is rather confusing. Originally, murine Sbsn-1 isoform was identified as a 700 aa long peptide [1]. A shorter isoform, lacking the longest isoform-specific internal repeats, is a putative Sbsn-2 (Q8CIT9-3; 164 aa; predicted mass 16.967 Da), which corresponds to human SBSN isoform-3 according to sequence analysis. Besides these two isoforms, Ensembl (version 101) identifies two other protein-coding isoforms supported by one or no EST, respectively. The UniProtKB database refers to six mouse Sbsn isoforms, including Sbsn-1 and Sbsn-2, the other isoforms are either duplications of Sbsn-1 or Sbsn-2, or unreviewed isoforms.
Secreted murine Sbsn-2 was detected using overexpression experiments [7], showing a mobility shift on immunoblots which indicates a post-translational modification. Indeed, murine Sbsn-1 is a substrate to tissue transglutaminase (Tgm) 2 and epithelial Tgm3 resulting in intermolecular crosslinking, and this represents the only verified post-translational modification in vitro. Its physiological relevance is still undetected. Citrullination of Sbsn was observed in the blood proteome of Parkinson’s disease in a rat model of pre-motor Parkinson’s disease [8]. Additionally, glycosylation of SBSN-1 was predicted [7] and indeed, using mass spectrometry approach, threonine 59 of human SBSN was identified to undergo O-glycosylation in various human cell lines [9]. Three protein kinase C (PKC) phosphorylation sites in C-terminus and two casein kinase II phosphorylation sites were predicted together with 73 potential N-myristoylation sites in the mouse homolog [1]; however, these predictions require experimental validation.
Three-dimensional structure of SBSN is still unknown. We performed Phyre2-based structure prediction [10] of all three human SBSN isoforms. In the case of SBSN-1, the predicted model predominantly consisted of disordered regions. Crystal structure of d337a mutant of Pseudomonas sp. mis38 lipase (PDB: 2ZJ6) served as a template for structure prediction with 12% sequence identity and 99.9% prediction confidence. Collagen α chain was among other possible templates with slightly lower confidence levels. The methyl-accepting chemotaxis protein of Escherichia coli (PDB: 1QU7), showing 13% sequence identity with SBSN-2, was used as a template for the structure prediction of the isoform with 98.5% confidence. Interestingly, the methyl-accepting chemotaxis protein of Thermotoga maritima (PDB: 2CH7) was suggested as a second template with 98.4% confidence. SBSN-3 isoform model was predicted upon the structure of human micelle-bound α-synuclein (PDB: 1XQ8), which showed 18% sequence identity.
4. Regulation of SBSN Expression
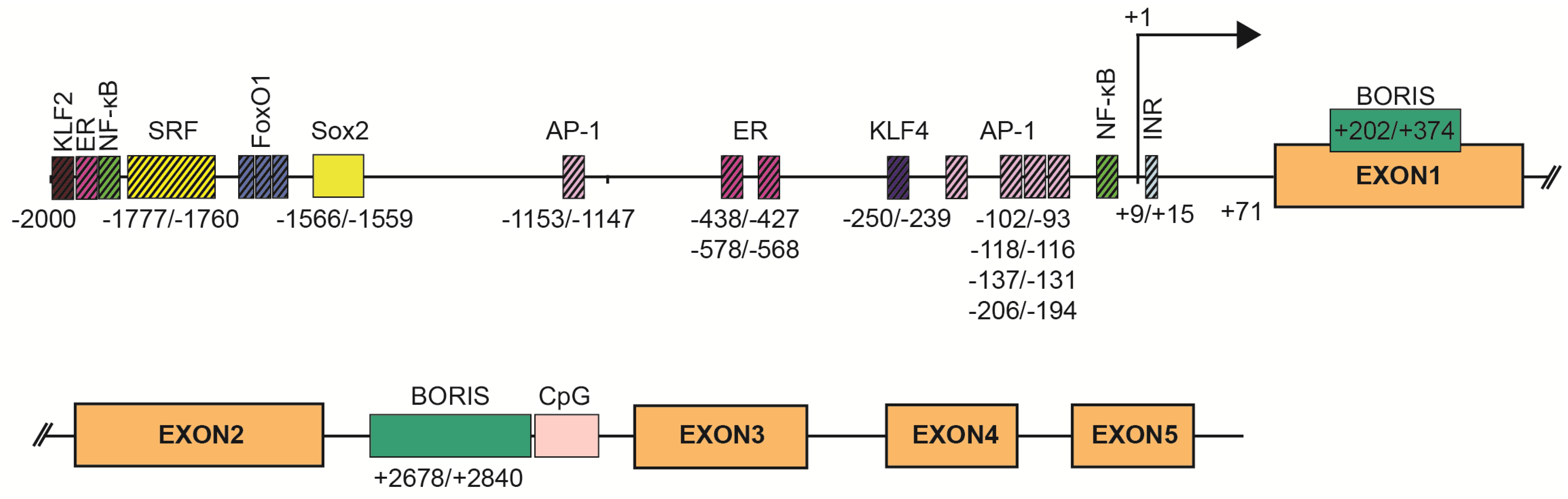
SBSN expression is tightly associated with stratified epithelia, but other expression sites have also been defined. Mechanisms of SBSN transcriptional control are not clearly described. We utilized the JASPAR algorithm [11] to predict binding sites of transcription factors in human (Supplementary Table S1) and mouse (Supplementary Table S2) SBSN proximal promoter (2 kbp upstream) and downstream coding region. Additionally, we verified putative binding sites. Based on these results, we established a model of human SBSN promoter (Figure 3). From transcription binding sites predicted within human SBSN proximal promoter region by in silico analysis, the SOX2 binding site is currently the only one experimentally validated [12] (Figure 3). Next, a Brother of the Regulator of Imprinted Sites (BORIS)-binding site within the coding region was experimentally verified [13]. Importantly, aberrant changes in expression of SBSN isoform are associated with atopic dermatitis (see below [14]), but mechanisms responsible for differential expression of isoforms under physiological and pathological conditions are not known.
SBSN is physiologically expressed in mouse stratified epithelia including the suprabasal epithelial layer of epidermis, tongue, oesophagus, palate, stomach, uterus, thyroid, trachea, lung, vagina, thymus, and urinary tract. In human, SBSN expression is associated with epidermis, thymus, uterus, tonsils, vagina, and oesophagus [1,2,15]. Originally, there was no evidence of SBSN expression in mouse embryonic and adult brain [1,7,16,17], but recent studies showed, using immunofluorescence, SBSN expression in human astrocytes, and its elevation under pathological conditions [18]. Transcriptome analysis of the human brain revealed SBSN mRNA levels in basal ganglia, but evaluation of SBSN protein presence in the human brain is needed to confirm this expression site [15]. On the contrary, the mouse brain does not show the presence of Sbsn mRNA; neither do several other mouse tissues and organs such as heart, kidney, or smooth muscle [1]. Therefore, between mouse and human, the SBSN expression sites seem to be conserved, except for the brain.
Mouse Sbsn mRNA was detected on day seven of embryonic development when the expression is likely mediated by extra-embryonic tissue. Hence, the embryonic expression was detected on day 15 of the development at first, which is in coincidence with epidermal stratification. Sbsn mRNA levels then peak on day 17 [1], and SBSN expression is associated with expression of dermokine-α/β [2], the components of secreted peptides complex. Similarly, SBSN mRNA was elevated in skeletal muscle cells of Alaskan sled dogs during an acute response (2 h post-exercise) after a prolonged endurance training together with dermokine and keratin 5 [17]. At the same time point, transcripts of genes involved in inflammation, oxidative stress, intermediary metabolism, immune response, and cellular compromise transcripts, e.g., S100A8, were also upregulated. The role of inflammation, immune, and stress response in SBSN expression is supported by the microarray analysis of therapy-resistant cancer cells in vitro, which showed transcript elevation of innate immune response genes and SBSN following 5-aza-2′-deoxycytidine (5-AC)-treatment or γ-radiation. Notably, activation of the ERK pathway downstream of IFN signalling emerged as a direct activator of SBSN expression [19]. Therefore, epidermal differentiation and response to inflammation provide hints at the understanding of SBSN expression inducing processes.
The composition of transcription factors and stimuli responsible for SBSN transcription is not specifically defined, but some are suggested. The Sbsn transcript elevates in differentiating mouse keratinocytes in vitro, whereas, several genes of the cornified envelope are downregulated upon SBSN knockdown [14]. This provides additional support for the coordinated gene expression program during skin differentiation [14]. Targets of the ERK pathway, i.e., components of the AP-1 transcription factor complex c-FOS, FRA-1, FRA-2, c-JUN, JUND, and JUNB, are differentially expressed in keratinocytes during their terminal differentiation in organotypic cultures, and AP-1 proteins are differentially expressed in the human epidermis [20]. This supports the role of ERK in SBSN expression; however, the role of ERK in keratinocyte differentiation provides contradictory results [21,22]. Additionally, MAL/SRF signalling also results in JUNB elevation, which plays an essential role in epidermal differentiation [21]. Inhibition of BCR-RHOA-MAL/SRF pathway resulted in the reduction of JUNB and SBSN transcripts, together with disruption of keratinocyte granulation and development of stratum corneum in an organotypic model of the human epidermis [23]. Mouse bearing conditional knockout of Srf in basal cells, showed reduced Sbsn transcript levels [24], and indeed, the SRF binding site was predicted within the SBSN proximal promoter region of both, human and mouse (with three binding sites in mice (−1994/−1978, −1036/−1023, −789/−773 (0.83, 0.8, and 0.8 relative score) and one binding site (−1777/−1760) in human with a 0.82 relative score). Changes in the actin cytoskeleton and MAL/SRF promote physical stimuli-induced keratinocyte differentiation via JUNB [21]. Note, in dog muscle, the JUNB transcript is elevated 2 h post-exercise, while the Fos transcript is downregulated [17]. ERK-mediated regulation of SBSN is further supported by increased expression of Sbsn in murine endothelial cells following treatment with Egf, but not bFgf [25]. Furthermore, phorbol 12-myristate 13-acetate (PMA)-mediated ERK activation enhanced SBSN expression efficiently [1,19], though this effect might be partially mediated by PKC since calcium-induced SBSN expression during differentiation of primary epidermal keratinocytes in vitro was suppressed with PKC inhibitor [1,2]. Therefore, multiple pathways activated during keratinocytes differentiation may promote SBSN expression likely via, but not only, AP-1-enabled transcription. Indeed, SBSN promoter region contains multiple AP-1 binding sites. In total, a JASPAR search predicted 72 AP-1 binding sites, with extensive sequence overlaps, hence lesser number of regions is more likely. Five regions showed >0.9 relative scores (−93/−102, −112/−118, −131/−137, −194/−206, −1147/−1153).
Lower temperature (33 °C) is frequently used in biotechnology for culture/propagation of Chinese hamster ovary cells. In a recent study, a list of cold-induced genes was established using RNAseq 48 h post change of condition, which included SBSN [26]. This was further supported with ectopic expression of luciferase driven by SBSN promoter exposed to lower temperatures. S100A4 was identified to be a cold-induced gene [26], together with S100A6, which is supported by a previous study [27]. Furthermore, the JunD binding site was predicted among selected cold-regulated promoters, including SBSN, and JUND transcript levels were elevated following cold-treatment [26].
In response to hypoxic stress, high altitude acclimatization leads to lower oxygen tension and hypobaric pressure with enhanced hematopoiesis, increased blood volume, and neoangiogenesis to redistribute blood flow to vital organs, including the brain. The latter is mediated by carotid arteries. Upon acclimatization to high-altitude-associated long-term hypoxia, SBSN transcript was one of 58 significantly upregulated in carotid arteries in sheep [28]. Interestingly, most altered genes were associated with cell migration, growth and proliferation, and angiogenesis. The authors also noted that some of the regulated genes are also common targets of treatment with lipopolysaccharide (LPS), again supporting the contribution of the innate immune response to SBSN expression. Note, the upstream pathway, ERK, and a target of SBSN signalling, AKT, showed increased activation in sheep carotid arteries accompanied long-term hypoxia [28].
The proximal region of SBSN gene promoter was originally described as AT-rich, lacking CpG islands and containing a canonical TATA box [1], however, in our analysis presented here (Figure 3) we were not able to identify these regions within 250 bp upstream of +1 site of human SBSN promoter region. Conversely, we observed an initiator element at +9/+15 (Figure 3). Multiple NF-kappaB binding sites were predicted within a 2 kbp region upstream of SBSN transcription start site in both human and mouse cells [29]. We predicted thirteen NF-kappaB binding sites within this region (Figure 3). The most prominent sites depicted had a relative score > 0.92; −1921/1930, > 0.8; −48/−39). Note, both PMA and LPS are potent inducers of NF-kappaB pathway. Furthermore, several other binding sites for transcription factors, such as SP1, TF2APA, MYC, SMAD2, and FOXO1/FOXO4 were predicted within the SBSN proximal promoter [1,30]. Indeed, SBSN transcription in confluent human adipocyte tissue-derived stem cells (ASCs) is mediated by FOXO1 [30], since downregulation of FOXO1 with silencing RNA reduced SBSN transcript levels. We predicted four FOXO1 binding sites within 2 kbp SBSN promoter region (relative score > 0.84; −1616/1609, −1668/−1661, −1692/−1685, −1930/−1923; Figure 3). Interestingly, SBSN promotes aromatase expression, and SBSN was shown to be induced with 17beta-estradiol treatment [31]. This is supported with the prediction of estrogen receptor (ER) binding sites within the promoter. We identified thirteen ER binding sites within 2 kbp upstream region of SBSN promoter and selected three highest scoring regions to depict (relative score > 0.89; −438/−427, > 0.86; −578/−568, > 0.85; −1902/−1888; Figure 3). Altogether, dozens of transcription factors binding sites are predicted. Needless to mention, confirmation of function of the predicted binding regions require further investigation.
Multiple studies described aberrant elevation of SBSN in human malignancies [19,29,32], but the mechanisms responsible for SBSN upregulation under pathological conditions is not understood. As mentioned, SBSN expression is limited to specific tissues. Bisulfite sequencing of 11 healthy human lung tissue samples revealed methylation of SBSN promoter CpG islands. This indicates that promoter methylation, and likely associated transcriptional repression, are responsible for SBSN silencing [33]. Indeed, normal human bronchial epithelial and human small airway epithelial cells treated with demethylating agent 5-AC and histone deacetylase inhibitor trichostatin A (TSA) elevated SBSN transcript levels [33]. This is further supported by hypomethylation of SBSN promoter in approximately 50% (13/28) of primary non-small cell lung carcinoma (NSCLC) samples observed in the same study. Hypomethylation of the SBSN promoter was an effect of a dysregulated proto-oncogenic zinc finger transcription factor, CTCFL/Brother of the Regulator of Imprinted Sites (BORIS) [13]. Similarly, SBSN transcription can be induced in vitro in a salivary gland adenoid cystic carcinoma (ACC) cell line with 5-AC and trichostatin A (TSA) resulting in hypomethylation of the CpG island [34]. The SBSN transcripts are induced with 5-AC in human cancer cell lines such as DU-145, MCF-7, and HeLa [19]. Notably, SBSN proteins were only detectable in a low-adherent subfraction of therapy-resistant 5-AC-treated cells with stem cell-like properties [19], indicating post-transcriptional regulation of SBSN expression. The SBSN gene promoter contains two experimentally confirmed CTCFL/BORIS binding sites in the first exon close to the transcription start site (+202/+374) and in the second intron in front of a CpG island (+2678/+2840). BORIS-mediated induction of SBSN is associated with demethylation of a CpG island in SBSN second intron and changes in histone marks comprising elevation of the active H3K4me3 and H3K14Ac, and downregulation of the repressive H3K9me3 modifications. Importantly, the elevation of SBSN transcript levels mediated by BORIS is dose-dependent. Relatively low BORIS levels were responsible for significantly higher SBSN transcript levels compared to high BORIS levels associated with re-methylation of the second intron of SBSN and increased nucleosome occupancy of SBSN transcription start site. A repressive histone mark H3K9me3 mirrored the SBSN second intron methylation pattern. Interestingly, CTCF and BORIS compete for the same binding sites of the SBSN promoter region [13], indicating that the epigenetic and chromatin state play essential roles in SBSN expression. Hence, methylation of regulatory sites represents the main feature responsible for the regulation (suppression) of SBSN transcription.
The connection between cell stemness and SBSN expression suggested previously [19] is supported by the identification of SOX2, a stem cell factor commonly upregulated in cancer, as a regulator of SBSN expression in oesophageal squamous cell carcinoma (ESCC) [12]. SOX2 binding site (−1566/−1559) at the proximal region of human SBSN was confirmed by chromatin immunoprecipitation (ChIP), and remains the only determining region of SBSN promoter with the validated transcription binding factor. Furthermore, double knockout Klf2 and Klf4 mouse cardiac microvascular endothelial cells showed significantly reduced SBSN transcript levels compared to control mice [35]. We identified 25 putative KLF4 binding sites and 30 putative KLF2 binding sites in the human SBSN promoter region, two most prominent are depicted (KLF4 relative score > 0.9; −250/−239, KLF2 relative score > 0.9; −1993/−1983; Figure 3). These observations strengthen the importance of factors of stemness in SBSN expression regulation.
5. The Function of SBSN in Context of Physiology and Pathology
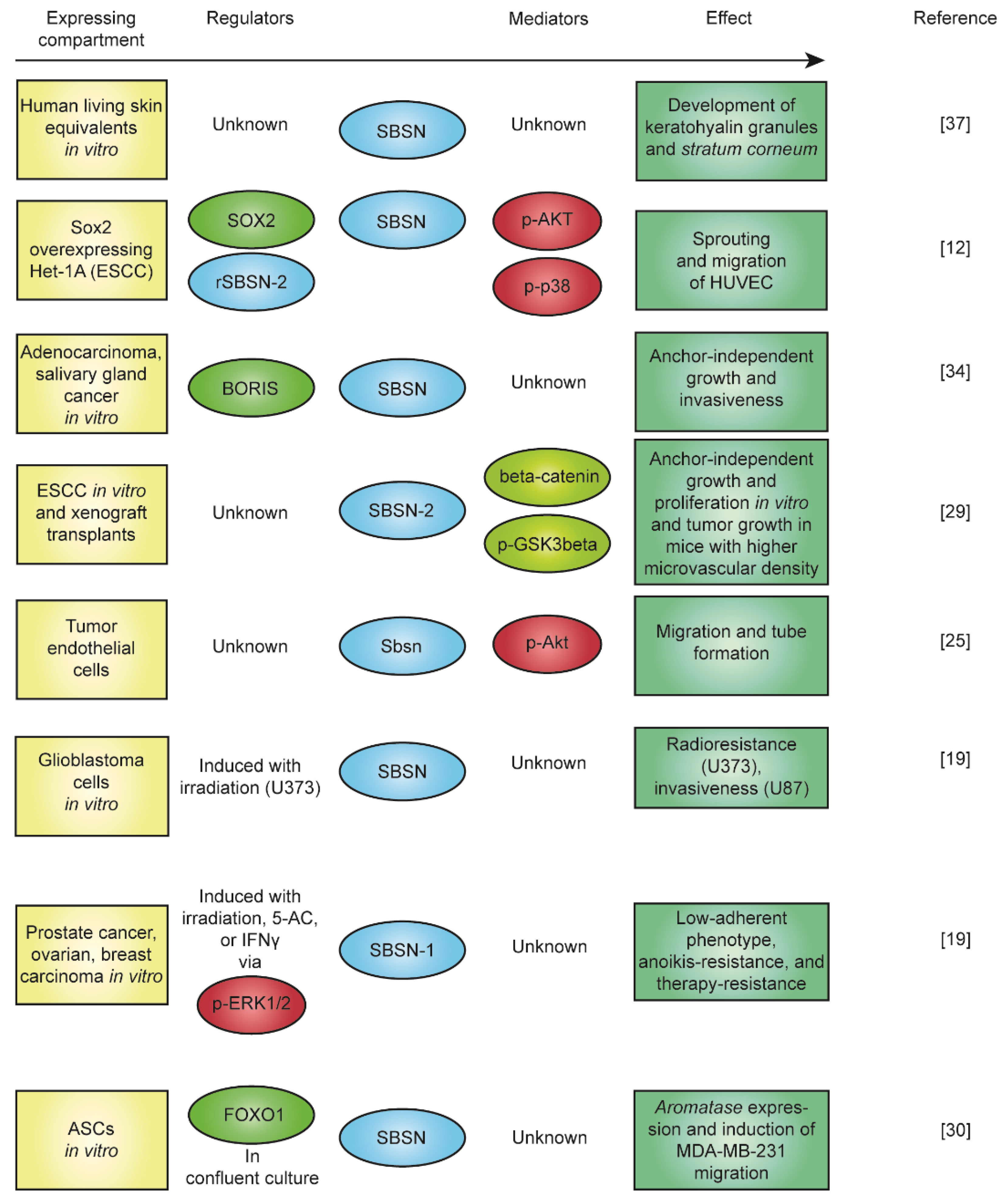
As already mentioned, the physiological function of SBSN is currently unknown. Sbsn knockout mice do not manifest any abnormal skin phenotype [36], but shRNA-mediated SBSN knockdown abrogated the development of stratum granulosum, disrupted the formation of keratohyalin granules, and affected the morphology of some keratinocytes in the human living skin equivalent model [37]. The secretory nature of SBSN isoforms is supported by a number of reports [2,7,32,38,39,40], and the SBSN treatment promoted phenotypic changes in vitro [12], indicating the existence of SBSN receptor(s). Studies focusing on the role of SBSN in human malignancies provided deeper insight regarding its function. Induction of SBSN activated AKT and p38MAPK kinases, and the WNT/β-catenin pathway [12,29]. The activation of WNT/β-catenin pathway was mediated specifically with overexpression of SBSN-2 [29]. And this served as a pro-angiogenic stimulus in mice [29]. Additionally, treatment with recombinant SBSN-2 enhanced sprouting in vitro [12]; thus, SBSN isoform 2 is a proposed oncogene in human malignancies. Therefore, SBSN can act as an oncogenic signalling molecule, but its receptor(s) and downstream signalling pathway(s) are currently unknown. The summary of the described functions of SBSN is depicted in Figure 4.
6. SBSN in Cancer
The most evidence about SBSN is from cancer research. Several studies have suggested the role of SBSN in adaptation to stress conditions [19], activation of pro-surviving signalling pathways [25,29], and angiogenesis [12,29], the features commonly associated with carcinogenesis. The amplification of the 19q13 region, which includes the SBSN coding sequence and its proximal promoter, is described in various cancer types, including ovarian, cervical, pancreatic, and breast carcinomas [41,42,43,44].
The first evidence of SBSN expression in cancer was a reported increase of SBSN mRNA levels linked with the hypomethylation of the SBSN promoter in almost half of non-small cell lung carcinoma cases [33]. The ectopic expression of SBSN in lung squamous cell lines resulted in increased anchorage-dependent growth in soft agar assay. The expression of SBSN in lung cancer correlates with BORIS [13,33]. BORIS is a transcription factor specific for the male germ line and is implicated in the activation of cancer-testis antigen (CTAs) genes [45,46,47]. BORIS is aberrantly expressed in several types of human cancers such as lung, head and neck, breast, and bladder carcinomas [48]. Additionally, SBSN CpG island in the second intron is significantly hypomethylated in primary salivary gland adenoid cystic carcinoma (ACC) compared to the normal salivary gland tissue. Knockdown of SBSN in a salivary gland ACC cell line suppressed anchorage-dependent growth in the soft agar and invasiveness in Matrigel invasion assay. Nevertheless, no correlation of SBSN with gender, smoking history, tumour or nodal stage, or metastatic disease of ACC was found. Also, no correlation with patient survival or time to disease relapse were found [34].
The oncogenic role of SBSN was originally proposed in a study of oesophageal squamous cell carcinoma (ESCC) [29]. In comparison to normal oesophageal epithelial cells, 11 ESCC cell lines showed elevated SBSN mRNA and the 25 kDa protein isoform. Importantly, ESCC tissue samples also showed elevated SBSN mRNA levels compared to unaffected tissue. Immunohistochemical analysis of 170 clinical ESCC specimens revealed a positive correlation between SBSN expression and tumour size, tumour clinical stage, and patient vital status. Patients with higher SBSN expression showed shorter overall survival [48]. A subsequent study suggested SBSN as a potential biomarker of ESCC [49]. The overexpression of SBSN isoform 2 promoted proliferation and anchorage-independent growth of ESCC cell lines in vitro (and normal human oesophageal epithelial cells as well) and tumour growth in vivo, whereas SBSN knockdown showed the opposite effect. SBSN-2 in ESCC cell lines increased the activity of WNT/β-catenin signalling pathway, which resulted in TCF/LEF transcriptional activity, nuclear translocation and reduced phosphorylation of WNT/β-catenin, and increased transcript levels of WNT/ β-catenin signalling-regulated genes such as AXIN2, MYC, CCND1, FRA1, MMP7 and JUN. The effect of SBSN on WNT signalling was mediated by phosphorylation of GSK3 β. WNT/ β-catenin signalling pathway regulates several cancer-associated processes such as cell growth and cell death, migration, invasiveness, stemness, and differentiation. Again, SBSN expressing ESCC cell lines formed in vivo tumours with higher microvascular density indicating the role of SBSN in angiogenesis [29]. In line with these observations, the treatment with recombinant SBSN-2 induced migration and sprouting of HUVEC [12]; however, this was accompanied by activation of AKT and p38MAPK. The phenotypic changes in the cells line were predominantly mediated by activated AKT since p38MAPK inhibition did not abrogate SBSN-2-promoted effects, whereas AKT inhibition did. Therefore, SOX2-regulated SBSN was suggested to mediate the angiogenic potential of early-stage ESCC via AKT signalling [12].
Tumour growth depends on de novo angiogenesis in tumour tissue. In accordance with the effects of SBSN in angiogenic processes [12,29], human tumour endothelial cells (TEC) isolated from renal cells and colon carcinomas expressed higher levels of SBSN compared to non-tumour (NEC) endothelial cells. SBSN was upregulated in mouse TEC isolated from human tumour xenografts, and its knockdown significantly suppressed VEGF-A-mediated migration and tube formation of mouse TEC compared to mouse NEC. Moreover, Sbsn knockdown reduced the phosphorylation status of Akt, but not Erk kinase, in mTEC indicates that Sbsn may take part in TEC migration, tube formation and pro-angiogenic activity via Akt activation [25]. The link of SBSN expression to vein cell function is further supported by an observation that knockdown of SBSN affected the capability of vein wall-derived cells to contract collagen gel in vitro [50].
In the case of malignant brain tumours, the higher SBSN transcript levels correlated with significantly lower survival of glioblastoma multiforme (GBM) patients. SBSN transcript levels are elevated in a GBM subtype with mesenchymal signature, the phenotype associated with a strong immunosuppressive milieu, therapy resistance, and the elevation of angiogenic markers [51,52]. Utilizing a mass spectrometric Stable isotope labelling with amino acids in cell culture (SILAC) approach, the secretome analysis of four glioblastoma cell lines (LN18, T98, U118 and U87; [38]) revealed SBSN peptides in the secretome of U87 together with other invasive factors. U87 was the most invasive cell line in a Matrigel invasion assay, but the role of SBSN was not determined. In another glioblastoma cell line U373, the siRNA-mediated knockdown of SBSN expression its participation in radioresistance via an unknown mechanism [19].
The role of SBSN in therapy resistance is supported by other findings. The expression of SBSN following radio- or chemo-therapy regimes in stem-like cells with a low-adhesive phenotype was mediated by ERK1/2 activity in breast, prostate and cervical cancer cell lines [19]. Knockdown of SBSN relieved ERK1/2 activity-dependent anoikis resistance of low-adherent cells. Similarities in a gene expression pattern in differentiating mouse skin [16] and irradiated low-adherent cells [19] can be observed. For instance, transcription factors Klf4, Klf6, Cebp α/β, Fos, Jun, JunB and Cdk inhibitor p21waf1 were upregulated during embryonic development of epidermis at days 14.5–15.5 and were detected in the transcriptome of low-adherent cancer cells as well. This indicates that the keratinocyte differentiation programme can take part in the development of low-adherent anoikis-resistant state of human cancer cells induced with genotoxic stress. The loss of adhesion, and, in some cases, nuclear degradation underscores the presence of keratinocyte differentiation-like regulatory network following therapy-induced phenotype.
SBSN was identified among 16 genes negatively associated with lymph node metastases in head and neck cancer (HNC) patients [53]. In this study, cornifelin (CNFN) represented a hub gene of the SBSN-containing gene cluster. This cluster was associated with gene set enriched for the p53 pathway and estrogen pathway genes. This suggests that the CNFN and SBSN-16-genes containing set could be functionally connected [53]. Previously, the estrogen biosynthesis and SBSN expression were linked via elevation of aromatase transcript levels in confluent ASCs [30] and induction of SBSN expression following 17beta-estradiol treatment of immortalized vaginal epithelial cells [31]. Importantly, SBSN in ASC-conditioned medium promoted migration of ER-negative breast carcinoma cell line MDA-MB-231 in an estrogen-independent manner [30]. Note, aberrantly elevated SBSN is observed in the bone marrow, and the peripheral blood of myelodysplastic syndromes (MDS) patients [32], hence systemic presence of SBSN accompany this disorder.
MDS represent a heterogeneous group of pre-leukemic diseases. MDS are characterized by ineffective haematopoiesis affecting the myeloid lineage predominantly [54]. Two studies showed the presence of SBSN in the blood of MDS patients. Firstly, SBSN was identified as an interactor of LRG1 derived from peripheral blood (PB) serum of MDS patients [55]. Note, LRG1, a granulocytes-progenitor receptor [56], was confirmed in neovascularization processes of retinal disease [57]. Secondly, SBSN expression was shown to be aberrantly elevated by myeloid compartment, predominantly by myeloid-derived suppressor cells (MDSCs) and early-stage MDSCs (eMDSCSs), in bone marrow (BM) of MDS patients [32]. Importantly, a subgroup of MDS patients with the highest bone marrow (BM) SBSN levels corresponded to a poor prognosis, high-risk group. SBSN expression was therapy-independent, and hence an intrinsic feature of the disease. Importantly, SBSN levels in peripheral blood reflected BM levels of SBSN [32]. Together with the observation of the presence of SBSN in the pleural fluid of patients suffering from lung adenocarcinoma [39], MDS represent another human malignancy in which SBSN is proposed as a potential biomarker [32]. Furthermore, SBSN aberrantly O-glycosylated at threonine 221 was observed specifically in peripheral blood (PB) plasma of gastric cancer patients [58] and reduced PB SBSN plasma levels in patients with atopic dermatitis [37] support that dysregulated SBSN plasma levels reflect disease conditions. Furthermore, BM SBSN showed a negative correlation with BM T cell counts and CCL2 levels in MDS, implicating anti-inflammatory conditions which might impede potentially the immunological response [32]. Importantly, SBSN-expressing BM MDSCs play a crucial role in the pathogenesis of MDS [59]; hence novel therapeutic strategies strive to target this cellular compartment [54]. Among multiple other factors, MDSCs-secreted alarmins S100A8/9 are mediators of the inflammatory response and subsequent pathogenicity mediated by MDSCs [60], this further supports the role of SBSN as a component of the innate immune response.
There is also limited evidence for a possible association of SBSN with ovarian carcinoma. Ovarian carcinoma derived SK-OV-3 cell line expresses SBSN in vitro and marked elevation of SBSN transcripts was found in patients’ biopsies of ovarian tumours [19]. These results indicate a potential function of SBSN in ovarian carcinoma, but the association of SBSN with the prognosis of ovarian cancer requires further investigation.
The relationship of SBSN expression with metastatic stages of malignant diseases is supported by a study employing mouse 4T1 cancer model. The blood-circulating 4T1 cells and 4T1 cells isolated from liver metastases showed higher Sbsn transcript levels compared to the primary tumour or parental 4T1 cells [19]. Furthermore, the link of Sbsn to metastatic disease is supported by another study [61]. To elucidate the role of T cell activity in the development of HNSCC, the microarray analysis of primary and metastatic sites in DMBA-induced HNSCC of nude and C57Bl6J mice were compared. The primary SCC in nude mice showed reduced transcript levels of terminal differentiation genes, such as Tgm3, desmoglein 1beta, and corneodesmosin specifically in nude mouse. Interestingly, histopathology revealed a difference in cervical lymph node metastatic tumours of wild-type and nude mice, showing reduced keratinization potential of these tumour sites developed in nude mice. This was accompanied with markedly reduced Sbsn transcript levels in metastatic SCC in nude mice. Comparison of primary and metastatic tumours cells developed in nude mice showed even stronger reduction in Sbsn expression [61]. Since T cell maturation-lacking nude mice showed markedly reduced expression of Sbsn in DMBA-induced HNSCC primary tumour cells and metastatic cells compared to C57Bl6J mice [61], we can speculate that Sbsn expression is a response of cancerous cells to anti-tumour T cell activity.
7. SBSN in Other Human Pathologies
With the onset of mass spectrometry (MS)-based proteomics, large datasets opened new avenues in understanding human diseases. Besides cancer, a number of studies showed the presence of SBSN peptides also under other pathological conditions, but only limited peptide counts and coverage of SBSN protein have been achieved [8,40,62]. The possibility of multiple post-translational modifications, and a therefore limited chance of identification, cannot be disqualified. Additionally, the combined analysis of SBSN mRNA and protein levels should be approached in subsequent studies.
To investigate the pathology of Fuchs endothelial corneal dystrophy (FECD), label-free quantitative tandem mass spectrometry proteome analysis was performed revealing SBSN peptides specifically in the aqueous humor of FECD patients [63]. FECD is an eye disorder causing corneal oedema and clouding, which results in vision impairment. FECD aetiology is unknown; however, the progressive loss of corneal endothelium is observed at the onset of the disease. Endothelial cell apoptosis likely promotes the synthesis of extracellular matrix (ECM) proteins responsible for tissue deformation and functional disruption [64]. Next to presence of SBSN peptides, downregulation of complement C3 protein and FAM3C (also downregulated in sporadic Alzheimer’s disease [65]) were identified as significant hits. Interestingly, spaceflight-exposed mouse model showed elevated Sbsn transcript levels in the retinal tissue [66], but this was not supported by later proteomic studies [67,68]. Similarly, the transcriptome analysis of cornea affected with fungal keratitis, a type of microbial infection which may eventually lead to loss of vision, revealed marked upregulation of anti-microbial, innate immune defence, wound healing, and cornified envelope genes. The latter included SPRR2A, SPRR2D, SPRR2F, SPRR3, and SBSN [69]. In line with described changes, several MMPs and pro-inflammatory cytokines were also elevated.
A follicular fluid isobaric tags for relative and absolute quantitation (iTRAQ) proteome study of polycystic ovary syndrome (PCOS) patients revealed downregulation of complement components and dysregulation of proteins functionally associated with angiogenic processes, for instance, PLG, AGT, LYVE1 (pro-angiogenic), and SERPINA1 (an inhibitor of PLG; anti-angiogenic). Importantly, the follicular fluid of PCOS patients was enriched in SBSN peptides [40]. Note, chronic inflammation, endothelial dysfunction, and elevated risk of cardiovascular disease, are commonly associated with PCOS [70,71,72].
In a rat model of pre-motor Parkinson’s disease (PD), the blood plasma proteome analysis of citrullinated proteins revealed a Sbsn peptide among PD-specific hits [8]. Citrullination is physiologically mediated by peptidylarginine deiminases (PAD). PAD gene expression has been shown in several organs but not in the thymus; therefore, citrullinated peptides are not subjected to central immune tolerance [73,74]. However, this may lead to neoantigen production and subsequent autoimmune recognition, the cause of potential autoreactivity. Pathological peptide citrullination is commonly associated with chronic inflammatory diseases and therefore considered as a response to chronic inflammation [74]. Citrullinated proteins recognized in other neurodegenerative diseases, including Huntington’s and Alzheimer’s diseases, suggest the role of aberrant peptide citrullination in the onset of these pathologies [73].
Another source of pathological citrullinated peptides is Porphyromonas gingivalis-expressed peptidylarginine deiminase (PPAD). Besides PPAD, P. gingivalis secretes a group of arginine-/lysine-targeting proteases, gingipains. Cleavage of host proteins with subsequent PPAD-mediated citrullination may lead to autoimmune response via antibodies recognizing the modified peptides. P. gingivalis is commonly associated with chronic periodontitis and oral squamous cell carcinoma [75]. Transient bacteremia may lead to transition of P. gingivalis to arteries [76], and arterial colonization with P. gingivalis is associated with cardiovascular diseases [77]. Additionally, P. gingivalis proteins are present in the majority of Alzheimer’s disease patients’ brains and correlate with the disease progression [78], and gingipains triggered Alzheimer-like disease phenotype in induced pluripotent stem cells-derived neurons [79]. Recently, Liu et al. treated oral squamous cancer cells with inactive P. gingivalis showing elevated SBSN mRNA levels 16 h post treatment. Additionally, tumour-associated macrophages showed pro-tumorigenic M2-transition following the bacterial treatment [75]. Altogether, these observations indicate that chronic inflammation and P. gingivalis infection could promote SBSN expression. Certainly, elucidation of the role of SBSN or presence of antibodies recognizing SBSN represent the next steps in further investigations.
The analysis of MHC II immunopeptidome revealed an abundance of SBSN peptides in axillary, brachial, inguinal, and skin-draining lymph nodes of healthy mice [62], indicating the importance of peripheral lymphoid organs in developing immune tolerance towards SBSN peptides, or posttranslationally modified SBSN peptides.
Indeed, in a recent search for neuropsychiatric systemic lupus erythematosus (NPSLE)-specific autoantibodies in cerebrospinal fluid (CSF) that could potentially be used as diagnostic markers of the disease, SBSN peptide-recognizing immune complexes were identified [18]. This indicates the presence of anti-SBSN autoantibodies specifically in a subgroup (nine out of 26 patients, only females) of NPSLE patients [18]. No SBSN immune complexes were found in the peripheral blood plasma of any NPSLE patients; therefore, the production of anti-SBSN autoantibodies by brain parenchyma-infiltrating lymphocytes intrathecally was suggested. However, the observation of SBSN immune complexes in CSF does not exclude the presence of anti-SBSN autoantibodies in the patients’ serum without the immune complexes formation. Additionally, authors showed higher SBSN expression in hippocampal astrocytes of NPSLE patients compared to healthy individuals where SBSN was also detected. Once again, these observations show aberrant SBSN expression under pathological conditions. Besides others, CSF of NPSLE patients is enriched in inflammatory cytokines [80], including IFN-gamma [81], a recently identified inducer of SBSN expression in cancer cells [19]. Interestingly, LPS-stimulated human normal astrocytes treated with commercially available anti-SBSN antibody showed elevated transcript levels of a few genes associated with senescence and autophagy, and TGF-beta signalling pathways [18].
8. SBSN, Bacterial Infection, Immune Response, and Obesity
As described above, SBSN transcript elevation follows P. gingivalis stimulation of oral cancer cells [75]. SBSN implication in bacterial response seems to be more profound, but yet not clear.
Trypanosoma cruzi represents a pathogen with an ability of congenital transmission. To elucidate the response of human prenatal tissue to T. cruzi infection, human placental extract (HPE) was infected with trypomastigotes of T. cruzi. Two hours post-treatment transcripts of pro-inflammatory cytokines, TLRs, and NLRs were elevated, but, 24 h post-infection, the SBSN transcript was among top upregulated genes [82]. Furthermore, mass spectrometry analysis of extracellular vesicles derived from human macrophages revealed SBSN peptides following LPS treatment (TLR4-stimuli; [83]). Additionally, human peripheral blood mononuclear cells (PBMC) treated with tRNA or 16 kDa lipoprotein (TLR1/2-stimulus) of Mycobacterium tuberculosis, or ssRNA (HIV-I derived sequence, TLR8-stimulus) significantly elevated SBSN mRNA levels after 6 h [84]. These results suggest SBSN expression can be a direct response following TLR receptors stimuli, and a downstream target of NF-kappaB as already proposed [29]; therefore SBSN is component of innate immune response and/or early anti-microbial defence.
Transcriptome analysis of vaginal epithelial cells from post-menopausal women with vaginal dryness, symptom commonly associated with vaginal microbial infection and epithelial irritation, showed downregulation of SBSN and TGM3 [85]. Gene ontology analysis revealed downregulation of cornified envelope, keratinocyte differentiation, immune response, and innate immune response genes in symptomatic women. In opposite, uropathogenic Escherichia coli (UPEC) colonizing the lumen and the epithelial cells of the bladder and responsible for chronic bacterial infection of the urinary tract, initiated innate immune defence and epithelial cell detachment [86,87]. Sbsn, Ivl, Lcn2, and complement components B and C3 were markedly elevated by murine urothelial cells in close proximity of intracellular UPEC colonies [88]. These results are supported by a subsequent in vitro study [31], in which the treatment of immortalised vaginal epithelial cells with 17beta-estradiol, flagellin of E. coli, or both, increased transcript levels of innate immune response, keratinization, and cell differentiation genes. LCN2, S100A8/9 (essential mediators of MDSC response in MDS [59]), SPRR2A and SPRR2F (essential in cornification processes [89]) were elevated with each treatment. SBSN together with CNFN, LCE3D, SPLI (cornification processes), and F3 were significantly elevated following 17beta-estradiol or 17beta-estradiol plus flagellin treatments. Besides others, 17β-estradiol elevated IL-17A transcript in the vaginal biopsies of treated post-menopausal women, implicating the stimulation of anti-microbial defence. Additionally, transcriptome analysis of mammary gland parenchyma of intact and ovariectomized prepubertal heifers revealed SBSN among estrogen-regulated genes [90].
Obesity is a comorbidity associated with multiple human diseases, including cancer. Chronic systemic inflammation underlies obesity-related symptoms [91]. In an attempt to elucidate the inflammatory role of human adipose tissue-derived stem cells (ASCs) under cell confluency in vitro, SBSN was found to be a FOXO1-regulated gene with a capacity to promote aromatase (CYP19A1 gene) expression. Similarly, FOXO1 depletion resulted in downregulation of CYP19A1 transcripts under confluent conditions. The ability of ASCs to express SBSN and aromatase was donor-dependent, hence likely dependent upon epigenetic state and priming of the cells [30]. The FOXO1-SBSN-aromatase pathway thus possibly results in elevation of estrogen levels, and as described above, 17beta-estradiol-induced SBSN transcription in vaginal epithelial cells. This implicates a positive feedback loop which could result in dysregulation of estrogen levels and SBSN-mediated response.
Despite its high prevalence, especially in children, atopic dermatitis (AD) remains a poorly understood and inefficiently treated immune system-driven cutaneous disease. Dysregulated immune cell-mediated cytokine signalling affecting the differentiation of epidermal cells as one of the features of AD is detrimental and predominantly driven by Th2-expressed IL-4 and IL-13 genes [92,93]. In the murine model, IL-4 was shown to downregulate the expression of epidermal barrier genes, Flg and Ivl [94]. Similarly, in vitro studies using NHEK revealed slight downregulation of SBSN mRNA following IL-4 treatments [14], but, human living skin equivalents did not reproduce these results following IL-4 treatment [37]. Healthy skin possesses mostly transcripts of SBSN isoform one, whereas levels of transcripts of isoform two and three are lower [14]. Interestingly, transcripts of all SBSN isoforms were markedly elevated in non-lesional skin samples of AD patients, but transcripts of all isoforms were reduced in skin lesion samples of AD patients [14]. The proteome analysis of skin tissue revealed a significant reduction of SBSN in AD patients as compared to healthy tissue. The lesional skin of AD patients showed varying SBSN staining intensities, including cases with low SBSN level staining as well as cases with SBSN intensities comparable to healthy tissue. Furthermore, AD and psoriasis vulgaris patients were characterized with reduced SBSN blood plasma levels, and patients with intrinsic AD type (non-systemic response), showed even reduced SBSN serum levels compared to extrinsic AD type (systemic, high serum IgE). shRNA-mediated SBSN knockdown induced apoptosis of fraction of keratinocytes in human living skin equivalent model, and IL-4/IL-13-treatments enhanced the observed apoptotic features, indicating SBSN-mediated resistance to IL-4/IL-13-induced apoptosis in human keratinocytes [37].
9. Conclusions
SBSN remains a gene of unknown molecular function. SBSN proteins represent secretory molecules with the ability to activate the signalling cascades, but its receptor(s) are not yet identified. Based on current evidence, SBSN is associated with multiple human diseases, including cancer and autoimmune disorders. Various cellular compartments express SBSN in diseases, hence uncovering stimuli and conditions responsible for SBSN expression in different cell types would provide valuable knowledge. Several SBSN expression-inducing stimuli are already confirmed (such as PMA, EGF, γ-radiation, and IFN-gamma). Also, other stimuli lead to SBSN transcript elevation (endotoxins and inflammatory signals), but their SBSN-inducing mechanisms remain to be elucidated. SBSN expression is likely post-transcriptionally regulated, and SBSN undergoes multiple post-translation modifications, which suggests a complex nature of SBSN biology. Importantly, dysregulated SBSN expression and SBSN plasma level alterations mark malignant conditions, potentially promoting systemic effect and patients response to the therapy [30,32,37]. Therefore, SBSN possesses the potential to serve as a biomarker of various human diseases [32,37,39] which has already been proposed [32,39].
Several cellular sources of SBSN expression were documented. Cancer cells expressing SBSN display stem markers [12,19] and indeed, SOX2 is currently the only identified transcription factor known to promote SBSN expression specifically in ESCC [12]. Tumour endothelial cells express SBSN in colon cancer and the effect of SBSN on endothelium is indicated in several reports [12,25], suggesting SBSN might play a role in the response of endothelial cells to aberrant conditions. Chronic inflammatory signalling accompanies the onset of tumorigenesis, promotes stemness [95], and the development of MDSCs [96]. MDSCs are known to mitigate immune response efficiently. Interestingly, MDSC-expressed SBSN was negatively correlated with BM T cell abundance in MDS [32]; hence SBSN could contribute to immune evasion of cancerous cells. These observations indicate that tumour microenvironment is causal for the induction of SBSN expression in different cell types of the malignant landscape.
The role of SBSN in immune regulation is strengthened with recent observation in Sbsn-null mice [36]. This animal model showed no aberration in skin development. However, following the low-dose nickel challenge, the mice did not develop T regulatory cells (Tregs) in the spleen, suggesting that SBSN is crucial in Treg development induced with these stimuli [36]. Other possibilities, such as induction of Treg development in general or induction of anergy in the tumour microenvironment specifically need to be evaluated in future studies.
In search for SBSN-inducing stimuli, we compiled various observations, indicating that innate immune response is a likely candidate [84,85]. SBSN transcripts elevated following treatments with bacterial components or lysates in different animal models [84,85]. Other genes of the innate immune response are upregulated together with SBSN transcripts, for example, S100A family representatives. Innate immune response accompanies early stages of tumour development associated with microenvironment remodelling and a dysregulated immune response [97]. Note, MDSCs-driven aberrant innate immune response in MDS BM contributes to the disease pathogenesis [59].
The fundamentals of SBSN are not yet understood; therefore, SBSN represents an interesting target for further studies in both physiology and pathology. We predict that SBSN will emerge in other diseases as a candidate diagnostic and prognostic biomarker. Understanding its function is of great importance since SBSN likely plays a role in disease development.
10. Future Perspectives
There is a plethora of questions regarding SBSN biology that remains to be answered:
- (1)
- Is SBSN a ligand and an inducer of signalling pathway?
Although the molecular function of SBSN has not been identified, current findings of SBSN secreted nature and the effects in experiments in vitro using recombinant SBSN indicate that SBSN can function as a ligand via an unknown receptor. The interacting partners of SBSN were not studied specifically. LRG1 remains to be the only shown interactor in peripheral blood of MDS patients [55]. Furthermore, the mechanism of activation of signalling AKT, p38MAPK, and/or WNT/β-catenin pathways or other pathways should be explored.
- (2)
- What stimulates aberrant expression of SBSN under malignant conditions?
Several signals that could promote aberrant SBSN expression associated with human malignancies were reported. Some of these are already documented with a degree of certainty, such as EGF [25], IFN-gamma [19]; however, the exact molecular mechanisms remain to be investigated. Stemness seems to be an important factor in SBSN regulation [12,19]. Additionally, cancer therapy itself (e.g., γ-radiation or 5-AC) may promote SBSN expression [19]. Multiple reports showing SBSN transcript elevation should be broadened by uncovering the SBSN molecular role, for instance, in innate immune response [31,84,88], estrogen [30,90], and temperature alterations [26]. Importantly, the mechanisms of SBSN post-transcriptional regulation, as observed in therapy-resistant cancer cells [19], need to be elucidated.
- (3)
- What is the functional role of individual SBSN isoforms?
Different protein isoforms may possess diverse and contradictory functions, and this cannot be excluded in case of SBSN. The oncogenic potential of SBSN is linked to human SBSN-2 [12,29] and should not be extrapolated to other isoforms unless proved experimentally. Indeed, SBSN-1 was shown to be expressed in therapy-resistant non-cycling tumour cells [19]; therefore, it could be speculated that human SBSN-1 and -2 promote contradictory effects. Post-translational modification of the isoforms can also alter SBSN-mediated effect. Strikingly, rodents lost the equivalent of human SBSN isoform 2, therefore elucidating SBSN-2 function utilizing rodent models is restricted to ectopic expression of human SBSN-2.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4425/12/1/108/s1, Table S1: Predicted binding sites of transcription factors within 2 kbp upstream region of human SBSN. Supplementary. Table S2: Predicted binding sites of transcription factors within 2 kbp upstream region of murine SBSN.
Author Contributions
M.P. wrote and edited the article and created the figures and the tables. I.K. Edited and reviewed the article, Z.H. reviewed the literature, wrote, reviewed and edited the article. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Grant Agency of the Czech Republic (Projects No. 17-07635S and 19-10543S) and an Institutional Grant (Project No. RVO 68378050).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We are very grateful to Marketa Vancurova for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Aa | Amino acids |
| AGT | Angiotensinogen |
| AKT | Thymomas of AKR (AKV retrovirus) mice |
| AP-1 | Activator protein-1 |
| AXIN2 | Axis inhibition protein 2 |
| bFGF | Basic fibroblast growth factor |
| BORIS | Brother of the regulator of imprinted sites |
| CCL2 | C-C motif chemokine ligand 2 |
| CDK | Cyclin-dependent kinase |
| CEBP | CCAAT enhancer-binding protein |
| CpGs | Cytosine phosphate guanine sites |
| CTCFL | CCCTC-binding factor like |
| Da | Daltons |
| DMBA | 7,12-Dimethylbenz[a]anthracene |
| EGF | Epidermal growth factor |
| ERK | Extracellular signal-regulated kinase |
| EST | Expressed sequence tag |
| FOS | Finkel-Biskis-Jinkins murine osteosarcoma |
| FOXO | Forkhead box |
| FRA | Fos-related antigen |
| GSK3beta | Glycogen synthase kinase 3 β |
| HUVEC | Human umbilical vein endothelial cells |
| IFN | Interferon |
| IgE | Immunoglobulin E |
| IL | Interleukin |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| kbp | kilobase pair |
| kDa | kilodaltons |
| Kdap | Keratinocyte differentiation-associated protein |
| KLF | Kruppel like factor |
| LYVE1 | Lymphatic vessel endothelial hyaluronan receptor 1 |
| MAL/SRF | Megakaryoblastic leukaemia 1/ serum response factor |
| MAPK | Mitogen-activated protein kinase |
| MMP7 | Matrix metallopeptidase 7 |
| NF-kappaB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLR | Nod-like receptors |
| PKC | Protein kinase C |
| PLG | Plasminogen |
| PMA | Phorbol 12-myristate 13-acetate |
| SERPINA1 | Serpin family a member 1 |
| SILAC | Stable isotope labelling with amino acids in cell culture |
| SOX2 | SRY (sex determining region Y)-box 2 |
| SP1 | Specificity protein 1 |
| SPRR | Small proline rich protein |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TLR | Toll-like receptors |
| VEGF | Vascular endothelial growth factor |
| WNT | Wingless-type |
References
- Park, G.T.; Lim, S.E.; Jang, S.I.; Morasso, M.I. Suprabasin, a novel epidermal differentiation marker and potential cornified envelope precursor. J. Biol. Chem. 2002, 277, 45195–45202. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Hayashi-Kisumi, F.; Kinoshita, Y.; Katahira, S.; Morita, K.; Miyachi, Y.; Ono, Y.; Imai, T.; Tanigawa, Y.; Komiya, T.; et al. Identification of novel keratinocyte-secreted peptides dermokine-α/- β and a new stratified epithelium-secreted protein gene complex on human chromosome 19q13.1. Genomics 2004, 84, 384–397. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Clark, H.F. The Secreted Protein Discovery Initiative (SPDI), a Large-Scale Effort to Identify Novel Human Secreted and Transmembrane Proteins: A Bioinformatics Assessment. Genome Res. 2003, 13, 2265–2270. [Google Scholar] [CrossRef] [Green Version]
- Consortium, T.U. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, P.; Salois, P.; St-Amant, N.; Gaumond, M.H.; Lanctôt, C. Identification of a conserved cluster of skin-specific genes encoding secreted proteins. Gene 2004, 334, 123–131. [Google Scholar] [CrossRef]
- Sancandi, M.; Uysal-Onganer, P.; Kraev, I.; Mercer, A.; Lange, S. Protein deimination signatures in plasma and plasma-evs and protein deimination in the brain vasculature in a rat model of pre-motor parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 2743. [Google Scholar] [CrossRef] [Green Version]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.-B.G.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Michael, J.E.S. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2016, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Mathelier, A.; Zhao, X.; Zhang, A.W.; Parcy, F.; Worsley-Hunt, R.; Arenillas, D.J.; Buchman, S.; Chen, C.Y.; Chou, A.; Ienasescu, H.; et al. JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014, 42, 142–147. [Google Scholar] [CrossRef]
- Takahashi, K.; Asano, N.; Imatani, A.; Kondo, Y.; Saito, M.; Takeuchi, A.; Jin, X.; Saito, M.; Hatta, W.; Asanuma, K.; et al. Sox2 induces tumorigenesis and angiogenesis of early stage esophagealsquamous cell carcinoma through secretion of Suprabasin. Carcinogenesis 2020, 26, 1–15. [Google Scholar] [CrossRef]
- Gaykalova, D.; Vatapalli, R.; Glazer, C.A.; Bhan, S.; Shao, C.; Sidransky, D.; Ha, P.K.; Califano, J.A. Dose-Dependent Activation of Putative Oncogene SBSN by BORIS. PLoS ONE 2012, 7, e40389. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zheng, L.; Uchiyama, A.; Bin, L.; Mauro, T.M.; Elias, P.M.; Pawelczyk, T.; Sakowicz-Burkiewicz, M.; Trzeciak, M.; Leung, D.Y.M.; et al. A data mining paradigm for identifying key factors in biological processes using gene expression data. Sci. Rep. 2018, 8, 9083. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Bazzi, H.; Fantauzzo, K.A.; Richardson, G.D.; Jahoda, C.A.B.; Christiano, A.M. Transcriptional profiling of developing mouse epidermis reveals novel patterns of coordinated gene expression. Dev. Dyn. 2007, 236, 961–970. [Google Scholar] [CrossRef] [Green Version]
- Brass, E.P.; Peters, M.A.; Hinchcliff, K.W.; He, Y.D.; Ulrich, R.G. Temporal pattern of skeletal muscle gene expression following endurance exercise in Alaskan sled dogs. J. Appl. Physiol. 2009, 107, 605–612. [Google Scholar] [CrossRef]
- Ichinose, K.; Ohyama, K.; Furukawa, K.; Higuchi, O.; Mukaino, A.; Satoh, K.; Nakane, S.; Shimizu, T.; Umeda, M.; Fukui, S.; et al. Novel anti-suprabasin antibodies may contribute to the pathogenesis of neuropsychiatric systemic lupus erythematosus. Clin. Immunol. 2018, 193, 123–130. [Google Scholar] [CrossRef]
- Hubackova, S.; Pribyl, M.; Kyjacova, L.; Moudra, A.; Dzijak, R.; Salovska, B.; Strnad, H.; Tambor, V.; Imrichova, T.; Svec, J.; et al. Interferon-regulated suprabasin is essential for stress-induced stem-like cell conversion and therapy resistance of human malignancies. Mol. Oncol. 2019, 13, 1467–1489. [Google Scholar] [CrossRef] [Green Version]
- Mehic, D.; Bakiri, L.; Ghannadan, M.; Wagner, E.F.; Tschachler, E. Fos and Jun Proteins Are Specifically Expressed During Differentiation of Human Keratinocytes. J. Investig. Dermatol. 2005, 124, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Connelly, J.T.; Gautrot, J.E.; Trappmann, B.; Tan, D.W.-M.; Donati, G.; Huck, W.T.S.; Watt, F.M. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat. Cell Biol. 2010, 12, 711–718. [Google Scholar] [CrossRef]
- Zhu, A.J.; Watt, F.M. beta-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development 1999, 126, 2285–2298. [Google Scholar]
- Dubash, A.D.; Koetsier, J.L.; Amargo, E.V.; Najor, N.A.; Harmon, R.M.; Green, K.J. The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein-1. J. Cell Biol. 2013, 202, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luxenburg, C.; Pasolli, H.A.; Williams, S.E.; Fuchs, E. Developmental roles for Srf, cortical cytoskeleton and cell shape in epidermal spindle orientation. Nat. Cell Biol. 2011, 13, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.T.; Nagao-Kitamoto, H.; Ohga, N.; Akiyama, K.; Maishi, N.; Kawamoto, T.; Shinohara, N.; Taketomi, A.; Shindoh, M.; Hida, Y.; et al. Suprabasin as a novel tumor endothelial cell marker. Cancer Sci. 2014, 105, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Novak, N.; Baumann, M.; Koehn, J.; Borth, N. Bioinformatic Identification of Chinese Hamster Ovary (CHO) Cold-Shock Genes and Biological Evidence of their Cold-Inducible Promoters. Biotechnol. J. 2019, e1900359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaisuchat, H.; Baumann, M.; Pontiller, J.; Hesse, F.; Ernst, W. Identification of a novel temperature sensitive promoter in cho cells. BMC Biotechnol. 2011, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, R.; Longo, L.D. Acclimatization to long-term hypoxia: Gene expression in ovine carotid arteries. Physiol. Genom. 2014, 46, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wu, G.; Li, Q.; Gong, H.; Song, J.; Cao, L.; Wu, S.; Song, L.; Jiang, L. Overexpression of Suprabasin is Associated with Proliferation and Tumorigenicity of Esophageal Squamous Cell Carcinoma. Sci. Rep. 2016, 6, 21549. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Dean, A.; Walter, M.; Bao, Y.; Hu, Y.; Ruan, J.; Li, R. Cell density-dependent transcriptional activation of endocrine-related genes in human adipose tissue-derived stem cells. Exp. Cell Res. 2010, 316, 2087–2098. [Google Scholar] [CrossRef] [Green Version]
- Stanton, A.; Mowbray, C.; Lanz, M.; Brown, K.; Hilton, P.; Tyson-Capper, A.; Pickard, R.S.; Ali, A.S.M.; Hall, J. Topical Estrogen Treatment Augments the Vaginal Response to Escherichia coli Flagellin. Sci. Rep. 2020, 10, 8473. [Google Scholar] [CrossRef]
- Pribyl, M.; Hubackova, S.; Moudra, A.; Vancurova, M.; Polackova, H.; Stopka, T.; Jonasova, A.; Bokorova, R.; Fuchs, O.; Stritesky, J.; et al. Aberrantly elevated suprabasin in the bone marrow as a candidate biomarker of advanced disease state in myelodysplastic syndromes. Mol. Oncol. 2020, 14, 2403–2419. [Google Scholar] [CrossRef]
- Glazer, C.A.; Smith, I.M.; Ochs, M.F.; Begum, S.; Westra, W.; Chang, S.S.; Sun, W.; Bhan, S.; Khan, Z.; Ahrendt, S.; et al. Integrative Discovery of Epigenetically Derepressed Cancer Testis Antigens in NSCLC. PLoS ONE 2009, 4, e8189. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Tan, M.; Bishop, J.A.; Liu, J.; Bai, W.; Gaykalova, D.A.; Ogawa, T.; Vikani, A.R.; Agrawal, Y.; Li, R.J.; et al. Suprabasin Is Hypomethylated and Associated with Metastasis in Salivary Adenoid Cystic Carcinoma. PLoS ONE 2012, 7, 1–7. [Google Scholar] [CrossRef]
- Sangwung, P.; Zhou, G.; Nayak, L.; Chan, E.R.; Kumar, S.; Kang, D.-W.; Zhang, R.; Liao, X.; Lu, Y.; Sugi, K.; et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017, 2, e91700. [Google Scholar] [CrossRef]
- Nakazawa, S.; Shimauchi, T.; Funakoshi, A.; Aoshima, M.; Phadungsaksawasdi, P.; Sakabe, J.; Asakawa, S.; Hirasawa, N.; Ito, T.; Tokura, Y. Suprabasin-null mice retain skin barrier function and show high contact hypersensitivity to nickel upon oral nickel loading. Sci. Rep. 2020, 10, 14559. [Google Scholar] [CrossRef]
- Aoshima, M.; Phadungsaksawasdi, P.; Nakazawa, S.; Iwasaki, M.; Sakabe, J.; Umayahara, T.; Yatagai, T.; Ikeya, S.; Shimauchi, T.; Tokura, Y. Decreased expression of suprabasin induces aberrant differentiation and apoptosis of epidermal keratinocytes: Possible role for atopic dermatitis. J. Dermatol. Sci. 2019, 95, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Formolo, C.A.; Williams, R.; Gordish-Dressman, H.; MacDonald, T.J.; Lee, N.H.; Hathout, Y. Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 2011, 10, 3149–3159. [Google Scholar] [CrossRef] [Green Version]
- Sheng, S.H.; Zhu, H.L. Proteomic analysis of pleural effusion from lung adenocarcinoma patients by shotgun strategy. Clin. Transl. Oncol. 2014, 16, 153–157. [Google Scholar] [CrossRef]
- Ambekar, A.S.; Kelkar, D.S.; Pinto, S.M.; Sharma, R.; Hinduja, I.; Zaveri, K.; Pandey, A.; Prasad, T.S.K.; Gowda, H.; Mukherjee, S. Proteomics of follicular fluid from women with polycystic ovary syndrome suggests molecular defects in follicular development. J. Clin. Endocrinol. Metab. 2015, 100, 744–753. [Google Scholar] [CrossRef]
- Kuuselo, R.; Simon, R.; Karhu, R.; Tennstedt, P.; Marx, A.H.; Izbicki, J.R.; Yekebas, E.; Sauter, G.; Kallioniemi, A. 19q13 amplification is associated with high grade and stage in pancreatic cancer. Genes. Chromosom. Cancer 2010, 49, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Thompson, F.H.; Nelson, M.A.; Trent, J.M.; Guan, X.-Y.; Liu, Y.; Yang, J.-M.; Emerson, J.; Adair, L.; Wymer, J.; Balfour, C.; et al. Amplification of 19q13.1–q13.2 sequences in ovarian cancer. Cancer Genet. Cytogenet. 1996, 87, 55–62. [Google Scholar] [CrossRef]
- Muleris, M.; Almeida, A.; Gerbault-Seureau, M.; Malfoy, B.; Dutrillaux, B. Identification of amplified DNA sequences in breast cancer and their organization within homogeneously staining regions. Genes Chromosom. Cancer 1995, 14, 155–163. [Google Scholar] [CrossRef]
- Rao, P.H.; Arias-Pulido, H.; Lu, X.-Y.; Harris, C.P.; Vargas, H.; Zhang, F.F.; Narayan, G.; Schneider, A.; Terry, M.B.; Murty, V.V. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer 2004, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.A.; Kang, Y.; Abdullaev, Z.; Flanagan, P.T.; Pack, S.D.; Fischette, M.R.; Adnani, M.T.; Loukinov, D.I.; Vatolin, S.; Risinger, J.I.; et al. Reciprocal Binding of CTCF and BORIS to the NY-ESO-1 Promoter Coincides with Derepression of this Cancer-Testis Gene in Lung Cancer Cells. Cancer Res. 2005, 65, 7763–7774. [Google Scholar] [CrossRef] [Green Version]
- Vatolin, S.; Abdullaev, Z.; Pack, S.D.; Flanagan, P.T.; Custer, M.; Loukinov, D.I.; Pugacheva, E.; Hong, J.A.; Morse, H.; Schrump, D.S.; et al. Conditional Expression of the CTCF-Paralogous Transcriptional Factor BORIS in Normal Cells Results in Demethylation and Derepression of MAGE-A1 and Reactivation of Other Cancer-Testis Genes. Cancer Res. 2005, 65, 7751–7762. [Google Scholar] [CrossRef] [Green Version]
- Bhan, S.; Negi, S.S.; Shao, C.; Glazer, C.A.; Chuang, A.; Gaykalova, D.A.; Sun, W.; Sidransky, D.; Ha, P.K.; Califano, J.A. BORIS Binding to the Promoters of Cancer Testis Antigens, MAGEA2, MAGEA3, and MAGEA4, Is Associated with Their Transcriptional Activation in Lung Cancer. Clin. Cancer Res. 2011, 17, 4267–4276. [Google Scholar] [CrossRef] [Green Version]
- Soltanian, S.; Dehghani, H. BORIS: A key regulator of cancer stemness. Cancer Cell Int. 2018, 18, 154. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, Q.; Su, Y.; Pan, L. Network-Based Differential Analysis to Identify Molecular Features of Tumorigenesis for Esophageal Squamous Carcinoma. Molecules 2018, 23, 88. [Google Scholar] [CrossRef] [Green Version]
- Kenagy, R.D.; Civelek, M.; Kikuchi, S.; Chen, L.; Grieff, A.; Sobel, M.; Lusis, A.J.; Clowes, A.W. Scavenger receptor class A member 5 (SCARA5) and suprabasin (SBSN) are hub genes of coexpression network modules associated with peripheral vein graft patency. J. Vasc. Surg. 2016, 64, 202–209.e6. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Huang, G.; Zhu, H.; Ma, Z.; Tian, X.; Yin, L.; Gao, X.; He, X. Analysis of gene co-expression network reveals prognostic significance of CNFN in patients with head and neck cancer. Oncol. Rep. 2019, 41, 2168–2180. [Google Scholar] [CrossRef]
- Winter, S.; Shoaie, S.; Kordasti, S.; Platzbecker, U. Integrating the “Immunome” in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef]
- Chrastinová, L.; Pastva, O.; Bocková, M.; Lynn, N.S.; Šácha, P.; Hubálek, M.; Suttnar, J.; Kotlín, R.; Štikarová, J.; Hlaváčková, A.; et al. A New Approach for the Diagnosis of Myelodysplastic Syndrome Subtypes Based on Protein Interaction Analysis. Sci. Rep. 2019, 9, 12647. [Google Scholar] [CrossRef] [Green Version]
- Druhan, L.J.; Lance, A.; Li, S.; Price, A.E.; Emerson, J.T.; Baxter, S.A.; Gerber, J.M.; Avalos, B.R. Leucine Rich α-2 Glycoprotein: A Novel Neutrophil Granule Protein and Modulator of Myelopoiesis. PLoS ONE 2017, 12, e0170261. [Google Scholar] [CrossRef]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Campos, D.; Freitas, D.; Gomes, J.; Magalhães, A.; Steentoft, C.; Gomes, C.; Vester-Christensen, M.B.; Ferreira, J.A.; Afonso, L.P.; Santos, L.L.; et al. Probing the O-Glycoproteome of Gastric Cancer Cell Lines for Biomarker Discovery. Mol. Cell. Proteom. 2015, 14, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Ku, T.K.S.; Crowe, D.L. Impaired T lymphocyte function increases tumorigenicity and decreases tumor latency in a mouse model of head and neck cancer. Int. J. Oncol. 2009, 35, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Fugmann, T.; Sofron, A.; Ritz, D.; Bootz, F.; Neri, D. The MHC Class II Immunopeptidome of Lymph Nodes in Health and in Chemically Induced Colitis. J. Immunol. 2017, 198, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Richardson, M.R.; Segu, Z.M.; Price, M.O.; Lai, X.; Witzmann, F.A.; Mechref, Y.; Yoder, M.C.; Price, F.W. Alterations in the aqueous humor proteome in patients with Fuchs endothelial corneal dystrophy. Mol. Vis. 2010, 16, 2376–2383. [Google Scholar]
- Nanda, G.G.; Alone, D.P. REVIEW: Current understanding of the pathogenesis of Fuchs’ endothelial corneal dystrophy. Mol. Vis. 2019, 25, 295–310. [Google Scholar]
- Liu, L.; Watanabe, N.; Akatsu, H.; Nishimura, M. Neuronal expression of ILEI/FAM3C and its reduction in Alzheimer’s disease. Neuroscience 2016, 330, 236–246. [Google Scholar] [CrossRef]
- Theriot, C.A.; Zanello, S.B. Molecular Effects of Spaceflight in the Mouse Eye after Space Shuttle Mission. Gravit. Space Res. 2014, 2, 3–24. [Google Scholar]
- Mao, X.; Byrum, S.; Nishiyama, N.; Pecaut, M.; Sridharan, V.; Boerma, M.; Tackett, A.; Shiba, D.; Shirakawa, M.; Takahashi, S.; et al. Impact of Spaceflight and Artificial Gravity on the Mouse Retina: Biochemical and Proteomic Analysis. Int. J. Mol. Sci. 2018, 19, 2546. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.W.; Nishiyama, N.C.; Byrum, S.D.; Stanbouly, S.; Jones, T.; Drew, A.; Sridharan, V.; Boerma, M.; Tackett, A.J.; Zawieja, D.; et al. Characterization of mouse ocular response to a 35-day spaceflight mission: Evidence of blood-retinal barrier disruption and ocular adaptations. Sci. Rep. 2019, 9, 8215. [Google Scholar] [CrossRef]
- Chidambaram, J.D.; Kannambath, S.; Srikanthi, P.; Shah, M.; Lalitha, P.; Elakkiya, S.; Bauer, J.; Prajna, N.V.; Holland, M.J.; Burton, M.J. Persistence of Innate Immune Pathways in Late Stage Human Bacterial and Fungal Keratitis: Results from a Comparative Transcriptome Analysis. Front. Cell. Infect. Microbiol. 2017, 7, 193. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Alexandraki, K.; Piperi, C.; Protogerou, A.; Katsikis, I.; Paterakis, T.; Lekakis, J.; Panidis, D. Inflammatory and endothelial markers in women with polycystic ovary syndrome. Eur. J. Clin. Investig. 2006, 36, 691–697. [Google Scholar] [CrossRef]
- Orio, F.; Palomba, S.; Spinelli, L.; Cascella, T.; Tauchmanovà, L.; Zullo, F.; Lombardi, G.; Colao, A. The Cardiovascular Risk of Young Women with Polycystic Ovary Syndrome: An Observational, Analytical, Prospective Case-Control Study. J. Clin. Endocrinol. Metab. 2004, 89, 3696–3701. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.C.J.; Lyall, H.; Petrie, J.R.; Gould, G.W.; Connell, J.M.C.; Sattar, N. Low Grade Chronic Inflammation in Women with Polycystic Ovarian Syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 2453–2455. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Maruyama, N. Importance of research on peptidylarginine deiminase and citrullinated proteins in age-related disease. Geriatr. Gerontol. Int. 2010, 10, S53–S58. [Google Scholar] [CrossRef] [PubMed]
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolino, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, X.; Peng, X.; Li, M.; Ren, B.; Cheng, G.; Cheng, L. Porphyromonas gingivalis Promotes Immunoevasion of Oral Cancer by Protecting Cancer from Macrophage Attack. J. Immunol. 2020, 205, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, J.; Mahendra, L.; Kurian, V.M.; Jaishankar, K.; Mythilli, R. Prevalence of periodontal pathogens in coronary atherosclerotic plaque of patients undergoing coronary artery bypass graft surgery. J. Maxillofac. Oral Surg. 2009, 8, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mougeot, J.-L.C.; Stevens, C.B.; Paster, B.J.; Brennan, M.T.; Lockhart, P.B.; Mougeot, F.K.B. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J. Oral Microbiol. 2017, 9, 1281562. [Google Scholar] [CrossRef] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Haditsch, U.; Roth, T.; Rodriguez, L.; Hancock, S.; Cecere, T.; Nguyen, M.; Arastu-Kapur, S.; Broce, S.; Raha, D.; Lynch, C.C.; et al. Alzheimer’s Disease-Like Neurodegeneration in Porphyromonas gingivalis Infected Neurons with Persistent Expression of Active Gingipains. J. Alzheimer’s Dis. 2020, 75, 1361–1376. [Google Scholar] [CrossRef]
- Yoshio, T.; Okamoto, H.; Kurasawa, K.; Dei, Y.; Hirohata, S.; Minota, S. IL-6, IL-8, IP-10, MCP-1 and G-CSF are significantly increased in cerebrospinal fluid but not in sera of patients with central neuropsychiatric lupus erythematosus. Lupus 2016, 25, 997–1003. [Google Scholar] [CrossRef]
- Wang, J.B.; Li, H.; Wang, L.L.; Liang, H.D.; Zhao, L.; Dong, J. Role of IL-1β, IL-6, IL-8 and IFN-γ in pathogenesis of central nervous system neuropsychiatric systemic lupus erythematous. Int. J. Clin. Exp. Med. 2015, 8, 16658–16663. [Google Scholar] [PubMed]
- Castillo, C.; Carrillo, I.; Libisch, G.; Juiz, N.; Schijman, A.; Robello, C.; Kemmerling, U. Host-parasite interaction: Changes in human placental gene expression induced by Trypanosoma cruzi. Parasit. Vectors 2018, 11, 479. [Google Scholar] [CrossRef]
- Lorey, M.B.; Rossi, K.; Eklund, K.K.; Nyman, T.A.; Matikainen, S. Global characterization of protein secretion from human macrophages following non-canonical caspase-4/5 inflammasome activation. Mol. Cell. Proteom. 2017, 16, S187–S199. [Google Scholar] [CrossRef] [Green Version]
- Keegan, C. Mycobacterium Tuberculosis tRNA Triggers TLR8 to Induce a Pathway for Th1 Cell Instruction. Ph.D. Thesis, University of California, Oakland, CA, USA, 2016. [Google Scholar]
- Hummelen, R.; Macklaim, J.M.; Bisanz, J.E.; Hammond, J.A.; McMillan, A.; Vongsa, R.; Koenig, D.; Gloor, G.B.; Reid, G. Vaginal microbiome and epithelial gene array in post-menopausal women with moderate to severe dryness. PLoS ONE 2011, 6, e26602. [Google Scholar] [CrossRef] [Green Version]
- Orikasa, S.; Hinman, F. Reaction of the vesical wall to bacterial penetration: Resistance to attachment, desquamation, and leukocytic activity. Investig. Urol. 1977, 15, 185–193. [Google Scholar]
- Mulvey, M.A. Induction and Evasion of Host Defenses by Type 1-Piliated Uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Hultgren, S.J.; Gordon, J.I. Functional genomic studies of uropathogenic Escherichia coli and host urothelial cells when intracellular bacterial communities are assembled. J. Biol. Chem. 2007, 282, 21259–21267. [Google Scholar] [CrossRef] [Green Version]
- Carregaro, F.; Stefanini, A.C.B.; Henrique, T.; Tajara, E.H. Study of small proline-rich proteins (SPRRs) in health and disease: A review of the literature. Arch. Dermatol. Res. 2013, 305, 857–866. [Google Scholar] [CrossRef]
- Li, R.W.; Meyer, M.J.; Van Tassell, C.P.; Sonstegard, T.S.; Connor, E.E.; Van Amburgh, M.E.; Boisclair, Y.R.; Capuco, A.V. Identification of estrogen-responsive genes in the parenchyma and fat pad of the bovine mammary gland by microarray analysis. Physiol. Genom. 2006, 27, 42–53. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y.M. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.D.; Suárez-Fariñas, M.; Dhingra, N.; Cardinale, I.; Li, X.; Kostic, A.; Ming, J.E.; Radin, A.R.; Krueger, J.G.; Graham, N.; et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Sehra, S.; Yao, Y.; Howell, M.D.; Nguyen, E.T.; Kansas, G.S.; Leung, D.Y.M.; Travers, J.B.; Kaplan, M.H. IL-4 Regulates Skin Homeostasis and the Predisposition toward Allergic Skin Inflammation. J. Immunol. 2010, 184, 3186–3190. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2009, 40, 22–35. [Google Scholar] [CrossRef]
- Gerada, C.; Ryan, K.M. Autophagy, the innate immune response and cancer. Mol. Oncol. 2020, 14, 1913–1929. [Google Scholar] [CrossRef]
Figure 1.
The gene tree of suprabasin SBSN. The gene tree of SBSN generated with MEGA X software [4] utilizing Enesembl (version 101) available data [3]. With the total sum of branch lengths (SBL) being >9.477. Distance scale = 0.10.

Figure 2.
The sequences of SBSN isoforms. (a) The amino acid sequences of human SBSN isoforms. (b) The gene organization of human SBSN isoforms.
Figure 2.
The sequences of SBSN isoforms. (a) The amino acid sequences of human SBSN isoforms. (b) The gene organization of human SBSN isoforms.

Figure 3.
SBSN promoter. Two kilobase pairs upstream of the promoter and downstream sequence of human SBSN with predicted transcription factor binding sites. SOX2 binding site is currently the only validated site [12] together with Brother of the Regulator of Imprinted Sites (BORIS) binding sites [13].
Figure 3.
SBSN promoter. Two kilobase pairs upstream of the promoter and downstream sequence of human SBSN with predicted transcription factor binding sites. SOX2 binding site is currently the only validated site [12] together with Brother of the Regulator of Imprinted Sites (BORIS) binding sites [13].

Figure 4.
SBSN and observed effects in various models. SBSN and observed effects based upon results from compiled literature. ESCC: oesophageal squamous cell carcinoma; HUVEC: human umbilical vein endothelial cells; ASCs: adipocyte tissue-derived stem cells.
Figure 4.
SBSN and observed effects in various models. SBSN and observed effects based upon results from compiled literature. ESCC: oesophageal squamous cell carcinoma; HUVEC: human umbilical vein endothelial cells; ASCs: adipocyte tissue-derived stem cells.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pribyl, M.; Hodny, Z.; Kubikova, I. Suprabasin—A Review. Genes 2021, 12, 108. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010108
AMA Style
Pribyl M, Hodny Z, Kubikova I. Suprabasin—A Review. Genes. 2021; 12(1):108. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010108
Chicago/Turabian StylePribyl, Miroslav, Zdenek Hodny, and Iva Kubikova. 2021. "Suprabasin—A Review" Genes 12, no. 1: 108. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010108
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.