Weighted Single-Step GWAS Identified Candidate Genes Associated with Growth Traits in a Duroc Pig Population

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals, Phenotypes, and Pedigree
2.3. Genotyping and Quality Control (QC)
2.4. Statistical Analyses
- Initially, set ;
- Calculate , where ;
- Calculate GEBVs for whole data set by ssGBLUP method;
- Calculate SNPs effects: , where was the GEBV of animals genotyped;
- Calculate the weight of each SNP:where was the -th SNP;
- Normalize SNP weights to keep total genetic variance constant via
- Set , then loop to step 2.
2.5. Identification of Candidate Genes and Functional Enrichment Analysis
3. Results and Discussion
3.1. Descriptive Statistics and Heritability for the Growth Traits
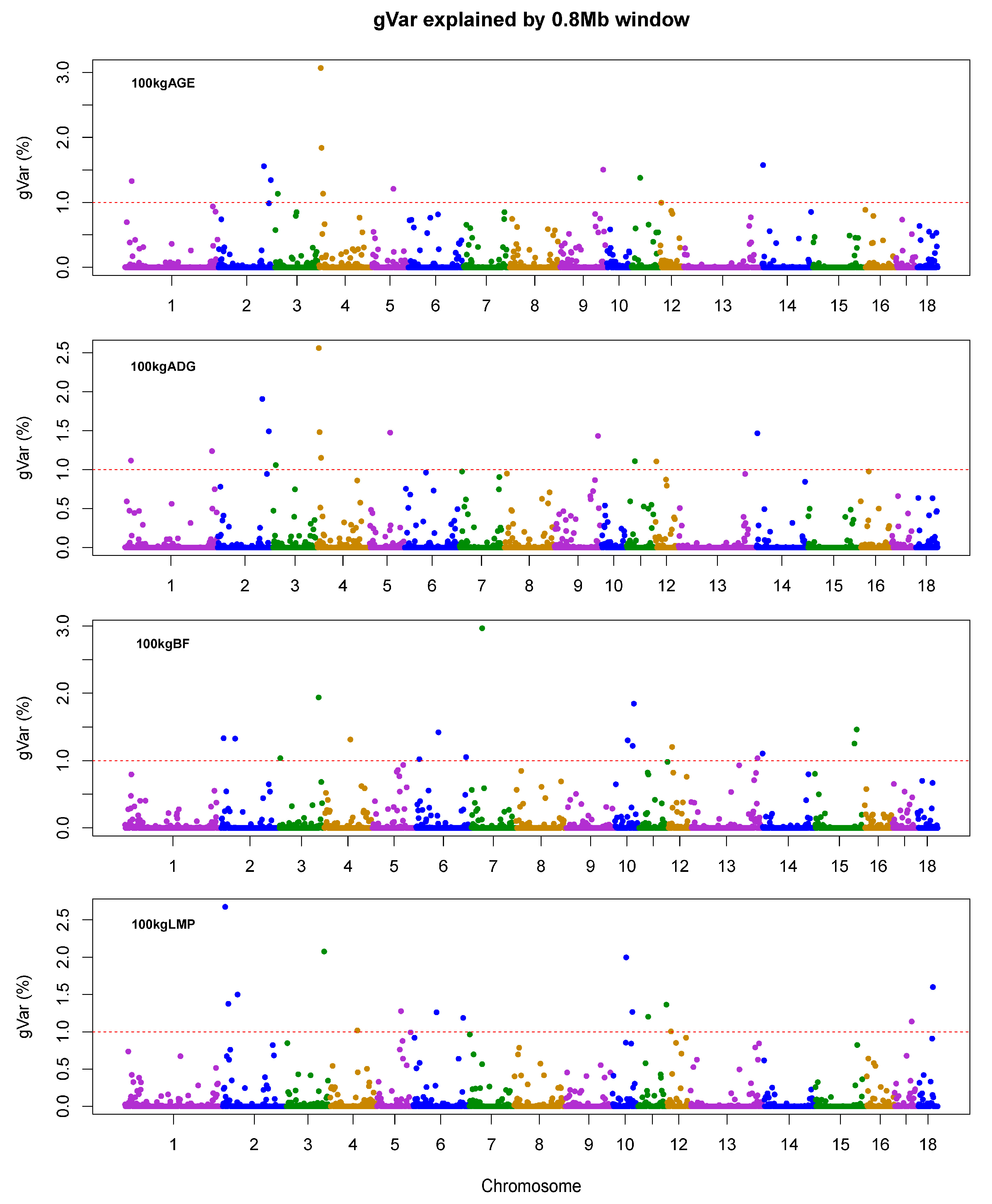
3.2. Summary of wssGWAS
3.3. wssGWAS for AGE and ADG
3.4. wssGWAS for BF
3.5. wssGWAS for LMP
3.6. BF and LMP Overlap Regions
3.7. GO and KEGG Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szűcs, I.; Vida, V. Global tendencies in pork meat-production, trade and consumption. Appl. Stud. Agribus. Commer. 2017, 11, 105–111. [Google Scholar] [CrossRef]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Russo, V. A genomewide association study for average daily gain in Italian Large White pigs. J. Anim. Sci. 2014, 92, 1385–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoque, M.A.; Suzuki, K.; Kadowaki, H.; Shibata, T.; Oikawa, T. Genetic parameters for feed efficiency traits and their relationships with growth and carcass traits in Duroc pigs. J. Anim. Breed. Genet. 2007, 124, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2018, 20, 135–156. [Google Scholar] [CrossRef]
- Jiang, Y.; Tang, S.; Wang, C.; Wang, Y.; Qin, Y.; Wang, Y.; Zhang, J.; Song, H.; Mi, S.; Yu, F.; et al. A genome-wide association study of growth and fatness traits in two pig populations with different genetic backgrounds. J. Anim. Sci. 2018, 96, 806–816. [Google Scholar] [CrossRef] [Green Version]
- Qiao, R.; Gao, J.; Zhang, Z.; Li, L.; Xie, X.; Fan, Y.; Cui, L.; Ma, J.; Ai, H.; Ren, J.; et al. Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet. Sel. Evol. 2015, 47, 17. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Fernando, R.L.; Vitezica, Z.; Okimoto, R.; Wing, T.; Hawken, R.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front. Genet. 2014, 5, 134. [Google Scholar] [CrossRef] [Green Version]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [Green Version]
- Stafuzza, N.B.; Silva, R.M.d.O.; Fragomeni, B.D.O.; Masuda, Y.; Huang, Y.J.; Gray, K.A.; Lourenco, D.A.L. A genome-wide single nucleotide polymorphism and copy number variation analysis for number of piglets born alive. Bmc Genom. 2019, 20, 321. [Google Scholar] [CrossRef]
- Silva, R.M.D.O.; Stafuzza, N.B.; Fragomeni, B.D.O.; De Camargo, G.M.F.; Ceacero, T.M.; Cyrillo, J.N.D.S.G.; Baldi, F.; Boligon, A.A.; Mercadante, M.E.Z.; Lourenco, D.L.; et al. Genome-Wide Association Study for Carcass Traits in an Experimental Nelore Cattle Population. PLoS ONE 2017, 12, e0169860. [Google Scholar] [CrossRef]
- Gonzalez-Pena, D.; Gao, G.; Baranski, M.; Moen, T.; Cleveland, B.M.; Kenney, P.B.; Vallejo, R.L.; Palti, Y.; Leeds, T.D. Genome-Wide Association Study for Identifying Loci that Affect Fillet Yield, Carcass, and Body Weight Traits in Rainbow Trout (Oncorhynchus mykiss). Front. Genet. 2016, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, N.; Chen, Y.; Liu, X.; Zhao, Y.; Zhu, L.; Liu, A.; Jiang, W.; Peng, X.; Zhang, C.; Tang, Z.; et al. Weighted single-step GWAS identified candidate genes associated with semen traits in a Duroc boar population. BMC Genom. 2019, 20, 797. [Google Scholar] [CrossRef]
- Tang, Z.; Xu, J.; Yin, L.; Yin, D.; Zhu, M.; Yu, M.; Li, X.; Zhao, S.; Liu, X. Genome-Wide Association Study Reveals Candidate Genes for Growth Relevant Traits in Pigs. Front. Genet. 2019, 10, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Kadowaki, H.; Shibata, T.; Uchida, H.; Nishida, A. Selection for daily gain, loin-eye area, backfat thickness and intramuscular fat based on desired gains over seven generations of Duroc pigs. Livest Prod. Sci. 2005, 97, 193–202. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, C.; Xuan, Y.; Zhou, P.; Fang, Z.; Che, L.; Xu, S.; Feng, B.; Li, J.; Jiang, X.; et al. Effect of maternal or post-weaning methyl donor supplementation on growth performance, carcass traits, and meat quality of pig offspring. J. Sci. Food Agric. 2019, 99, 2096–2107. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.R.; Sonstegard, T.S.; Schnabel, R.D.; Taylor, J.F.; Schenkel, F.S. Invited review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Z.; Ding, R.; Peng, L.; Wu, J.; Ye, Y.; Zhou, S.; Wang, X.; Quan, J.; Zheng, E.; Cai, G.; et al. Genome-wide association analyses identify known and novel loci for teat number in Duroc pigs using single-locus and multi-locus models. BMC Genom. 2020, 21, 344. [Google Scholar] [CrossRef] [PubMed]
- Misztal, I.; Tsuruta, S.; Strabel, T.; Auvray, B.; Druet, T.; Lee, D. BLUPF90 and related programs (BGF90). In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, 19–23 August 2002; pp. 743–744. [Google Scholar]
- Herrera-Cáceres, W.; Sánchez, J.P. Selection for feed efficiency using the social effects animal model in growing Duroc pigs: Evaluation by simulation. Genet. Sel. Evol. 2020, 52, 53. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.P.; Tribout, T.; Iannuccelli, N.; Bouffaud, M.; Servin, B.; Tenghe, A.; Dehais, P.; Muller, N.; Del Schneider, M.P.; Mercat, M.J.; et al. A genome-wide association study of production traits in a commercial population of Large White pigs: Evidence of haplotypes affecting meat quality. Genet. Sel. Evol. 2014, 46, 12. [Google Scholar] [CrossRef] [Green Version]
- van Kuijk, S.J.A.; Jacobs, M.; Smits, C.H.M.; Han, Y. The effect of hydroxychloride trace minerals on the growth performance and carcass quality of grower/finisher pigs: A meta-analysis. J. Anim. Sci. 2019, 97, 4619–4624. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.T.; Jiao, S.; Tiezzi, F.; Huang, Y.; Gray, K.A.; Maltecca, C. Genome-wide association study on legendre random regression coefficients for the growth and feed intake trajectory on Duroc Boars. BMC Genet. 2015, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Ødegård, J.; Meuwissen, T.H. Estimation of heritability from limited family data using genome-wide identity-by-descent sharing. Genet. Sel. Evol. 2012, 44, 16. [Google Scholar] [CrossRef] [Green Version]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimaraes, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50, 40. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.O.; Kizilkaya, K.; Garrick, D.J.; Fernando, R.L.; Reecy, J.M.; Weaber, R.L.; Silver, G.A.; Thomas, M.G. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J. Anim. Sci. 2012, 90, 3398–3409. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- Bergamaschi, M.; Maltecca, C.; Fix, J.; Schwab, C.; Tiezzi, F. Genome-wide association study for carcass quality traits and growth in purebred and crossbred pigs1. J. Anim. Sci. 2020, 98. [Google Scholar] [CrossRef]
- Yang, J.; Huang, L.; Yang, M.; Fan, Y.; Li, L.; Fang, S.; Deng, W.; Cui, L.; Zhang, Z.; Ai, H.; et al. Possible introgression of the VRTN mutation increasing vertebral number, carcass length and teat number from Chinese pigs into European pigs. Sci. Rep. 2016, 6, 19240. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Steibel, J.P.; Bates, R.O.; Raney, N.E.; Rumph, J.M.; Ernst, C.W. Identification of Carcass and Meat Quality QTL in an F(2) Duroc × Pietrain Pig Resource Population Using Different Least-Squares Analysis Models. Front. Genet. 2011, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Bennett, H.P. The granulin gene family: From cancer to dementia. BioEssays 2009, 31, 1245–1254. [Google Scholar] [CrossRef]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef] [Green Version]
- Bassett, J.H.; Williams, G.R. Role of Thyroid Hormones in Skeletal Development and Bone Maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kuang, Z.; Yu, X.; Ruhn, K.A.; Kubo, M.; Hooper, L.V. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science 2017, 357, 912–916. [Google Scholar] [CrossRef] [Green Version]
- DeChiara, T.M.; Kimble, R.B.; Poueymirou, W.T.; Rojas, J.; Masiakowski, P.; Valenzuela, D.M.; Yancopoulos, G.D. Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nat. Genet. 2000, 24, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Reyer, H.; Varley, P.F.; Murani, E.; Ponsuksili, S.; Wimmers, K. Genetics of body fat mass and related traits in a pig population selected for leanness. Sci. Rep. 2017, 7, 9118. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.G.; Gui, L.S.; Wang, H.C.; Tian, W.Q.; Li, Y.K.; Zan, L.S. Polymorphisms in adrenergic receptor genes in Qinchuan cattle show associations with selected carcass traits. Meat Sci. 2018, 135, 166–173. [Google Scholar] [CrossRef]
- Bachman, E.S.; Dhillon, H.; Zhang, C.Y.; Cinti, S.; Bianco, A.C.; Kobilka, B.K.; Lowell, B.B. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science 2002, 297, 843–845. [Google Scholar] [CrossRef] [Green Version]
- Odgren, P.R.; Kim, N.; MacKay, C.A.; Mason-Savas, A.; Choi, Y.; Marks, S.C., Jr. The role of RANKL (TRANCE/TNFSF11), a tumor necrosis factor family member, in skeletal development: Effects of gene knockout and transgenic rescue. Connect. Tissue Res. 2003, 44, 264–271. [Google Scholar] [CrossRef]
- Whyte, M.P.; Totty, W.G.; Novack, D.V.; Zhang, X.; Wenkert, D.; Mumm, S. Camurati-Engelmann disease: Unique variant featuring a novel mutation in TGFβ1 encoding transforming growth factor β 1 and a missense change in TNFSF11 encoding RANK ligand. J. Bone Miner. Res. 2011, 26, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Heemers, H.; Vanderhoydonc, F.; Roskams, T.; Shechter, I.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Androgens stimulate coordinated lipogenic gene expression in normal target tissues in vivo. Mol. Cell. Endocrinol. 2003, 205, 21–31. [Google Scholar] [CrossRef]
- Lin, D.Y.; Lai, M.Z.; Ann, D.K.; Shih, H.M. Promyelocytic leukemia protein (PML) functions as a glucocorticoid receptor co-activator by sequestering Daxx to the PML oncogenic domains (PODs) to enhance its transactivation potential. J. Biol. Chem. 2003, 278, 15958–15965. [Google Scholar] [CrossRef] [Green Version]
- Tordoff, M.G.; Ellis, H.T. Taste dysfunction in BTBR mice due to a mutation of Itpr3, the inositol triphosphate receptor 3 gene. Physiol. Genom. 2013, 45, 834–855. [Google Scholar] [CrossRef] [Green Version]
- Moritoh, Y.; Oka, M.; Yasuhara, Y.; Hozumi, H.; Iwachidow, K.; Fuse, H.; Tozawa, R. Inositol hexakisphosphate kinase 3 regulates metabolism and lifespan in mice. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Z.; Zhang, Z.; Xiao, Q.; Mawed, S.; Xu, Z.; Zhang, X.; Yang, H.; Zhu, M.; Xue, M.; et al. Genomic signatures reveal selection of characteristics within and between Meishan pig populations. Anim Genet. 2018, 49, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Li, S.; Ding, R.; Yang, M.; Zheng, E.; Yang, H.; Gu, T.; Xu, Z.; Cai, G.; Wu, Z.; et al. Meta-analysis of genome-wide association studies for loin muscle area and loin muscle depth in two Duroc pig populations. PLoS ONE 2019, 14, e0218263. [Google Scholar] [CrossRef]
- Li, T.; Chanda, D.; Zhang, Y.; Choi, H.S.; Chiang, J.Y. Glucose stimulates cholesterol 7alpha-hydroxylase gene transcription in human hepatocytes. J. Lipid Res. 2010, 51, 832–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrescu, A.M.; Liao, X.H.; Abdullah, M.S.; Lado-Abeal, J.; Majed, F.A.; Moeller, L.C.; Boran, G.; Schomburg, L.; Weiss, R.E.; Refetoff, S. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat. Genet. 2005, 37, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, D.; Zhang, F.; Tu, Y.; Xia, Y.; Wang, H.; Zhou, B.; Zhang, Y.; Wu, J.; Gao, X.; et al. Liver Patt1 deficiency protects male mice from age-associated but not high-fat diet-induced hepatic steatosis. J. Lipid Res. 2012, 53, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, Y.; Gong, H.; Cui, L.; Ma, J.; Chen, C.; Ai, H.; Xiao, S.; Huang, L.; Yang, B. Landscape of Loci and Candidate Genes for Muscle Fatty Acid Composition in Pigs Revealed by Multiple Population Association Analysis. Front. Genet. 2019, 10, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.-Y.; Yu, L.; Graham, J.; Hsu, D.K.; Lloyd, K.C.K.; Havel, P.J.; Liu, F.-T. Ablation of a galectin preferentially expressed in adipocytes increases lipolysis, reduces adiposity, and improves insulin sensitivity in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 18696–18701. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, D.; Yin, Y.; Ji, M.; Xu, K.; Huang, X.; Peng, Y.; Zhang, J. Comprehensive transcriptomic view of the role of the LGALS12 gene in porcine subcutaneous and intramuscular adipocytes. BMC Genom. 2019, 20, 509. [Google Scholar] [CrossRef]
- Seres, J.; Bornstein, S.R.; Seres, P.; Willenberg, H.S.; Schulte, K.M.; Scherbaum, W.A.; Ehrhart-Bornstein, M. Corticotropin-releasing hormone system in human adipose tissue. J. Clin. Endocrinol. Metab. 2004, 89, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Hofer, D.C.; Pessentheiner, A.R.; Pelzmann, H.J.; Schlager, S.; Madreiter-Sokolowski, C.T.; Kolb, D.; Eichmann, T.O.; Rechberger, G.; Bilban, M.; Graier, W.F.; et al. Critical role of the peroxisomal protein PEX16 in white adipocyte development and lipid homeostasis. Biochim. Et Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 358–368. [Google Scholar] [CrossRef]
- Mármol-Sánchez, E.; Quintanilla, R.; Cardoso, T.F.; Jordana Vidal, J.; Amills, M. Polymorphisms of the cryptochrome 2 and mitoguardin 2 genes are associated with the variation of lipid-related traits in Duroc pigs. Sci. Rep. 2019, 9, 9025. [Google Scholar] [CrossRef]
- Krause, K.; Weiner, J.; Hönes, S.; Klöting, N.; Rijntjes, E.; Heiker, J.T.; Gebhardt, C.; Köhrle, J.; Führer, D.; Steinhoff, K.; et al. The Effects of Thyroid Hormones on Gene Expression of Acyl-Coenzyme A Thioesterases in Adipose Tissue and Liver of Mice. Eur. Thyroid J. 2015, 4, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Godinho, R.M.; Bergsma, R.; Silva, F.F.; Sevillano, C.A.; Knol, E.F.; Lopes, M.S.; Lopes, P.S.; Bastiaansen, J.W.M.; Guimarães, S.E.F. Genetic correlations between feed efficiency traits, and growth performance and carcass traits in purebred and crossbred pigs. J. Anim. Sci. 2018, 96, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.; Tanaka, Y.; Takahashi, A.; Hirose, H.; Kashiwagi, A.; Kaku, K.; Kawamori, R.; Nakamura, Y.; Maeda, S. Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes 2008, 57, 791–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasza, I.; Suh, Y.; Wollny, D.; Clark, R.J.; Roopra, A.; Colman, R.J.; MacDougald, O.A.; Shedd, T.A.; Nelson, D.W.; Yen, M.I.; et al. Syndecan-1 is required to maintain intradermal fat and prevent cold stress. PLoS Genet. 2014, 10, e1004514. [Google Scholar] [CrossRef] [Green Version]
- Belaid, Z.; Hubint, F.; Humblet, C.; Boniver, J.; Nusgens, B.; Defresne, M.P. Differential expression of vascular endothelial growth factor and its receptors in hematopoietic and fatty bone marrow: Evidence that neuropilin-1 is produced by fat cells. Haematologica 2005, 90, 400–401. [Google Scholar]
- Dai, X.; Okon, I.; Liu, Z.; Bedarida, T.; Wang, Q.; Ramprasath, T.; Zhang, M.; Song, P.; Zou, M.H. Ablation of Neuropilin 1 in Myeloid Cells Exacerbates High-Fat Diet-Induced Insulin Resistance Through Nlrp3 Inflammasome In Vivo. Diabetes 2017, 66, 2424–2435. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.G.; Kenny, D.A.; Fitzsimons, C.; Blackshields, G.; Coyle, S.; McKenna, C.; McGee, M.; Morris, D.W.; Waters, S.M. The effect of breed and diet type on the global transcriptome of hepatic tissue in beef cattle divergent for feed efficiency. BMC Genom. 2019, 20, 525. [Google Scholar] [CrossRef]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Bhoj, E.J.; Romeo, S.; Baroni, M.G.; Bartov, G.; Schultz, R.A.; Zinn, A.R. MODY-like diabetes associated with an apparently balanced translocation: Possible involvement of MPP7 gene and cell polarity in the pathogenesis of diabetes. Mol. Cytogenet. 2009, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Shim, M.Y.; Karnuah, A.B.; Mitchell, A.D.; Anthony, N.B.; Pesti, G.M.; Aggrey, S.E. The effects of growth rate on leg morphology and tibia breaking strength, mineral density, mineral content, and bone ash in broilers. Poult. Sci. 2012, 91, 1790–1795. [Google Scholar] [CrossRef]
- Cameron, N.D.; Curran, M.K. Responses in carcass composition to divergent selection for components of efficient lean growth rate in pigs. Anim. Sci. 2010, 61, 347–359. [Google Scholar] [CrossRef]
- Wang, W.; Xue, W.; Jin, B.; Zhang, X.; Ma, F.; Xu, X. Candidate gene expression affects intramuscular fat content and fatty acid composition in pigs. J. Appl Genet. 2013, 54, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.; Cota, D.; Seeley, R.J. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu. Rev. Physiol. 2008, 70, 513–535. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Chang, R.B.; Bushman, J.D.; Tu, Y.H.; Mulhall, E.M.; Wilson, C.E.; Cooper, A.J.; Chick, W.S.; Hill-Eubanks, D.C.; Nelson, M.T.; et al. The K+ channel KIR2.1 functions in tandem with proton influx to mediate sour taste transduction. Proc. Natl. Acad. Sci. USA 2016, 113, E229–E238. [Google Scholar] [CrossRef] [Green Version]
- Ribani, A.; Bertolini, F.; Schiavo, G.; Scotti, E.; Utzeri, V.J.; Dall’Olio, S.; Trevisi, P.; Bosi, P.; Fontanesi, L. Next generation semiconductor based sequencing of bitter taste receptor genes in different pig populations and association analysis using a selective DNA pool-seq approach. Anim. Genet. 2017, 48, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Des Gachons, C.P.; Breslin, P.A. Salivary amylase: Digestion and metabolic syndrome. Curr. Diabetes Rep. 2016, 16, 102. [Google Scholar] [CrossRef]


{kind=link}
| Traits a | n | Mean | SD b | Min | Max | CV (%) c |
|---|---|---|---|---|---|---|
| AGE | 3927 | 163.41 | 11.93 | 125.98 | 206.32 | 7.30 |
| ADG | 3927 | 604.31 | 43.81 | 478.73 | 779.49 | 7.25 |
| BF | 3909 | 9.52 | 1.70 | 5.10 | 17.31 | 17.86 |
| LMP | 3909 | 61.08 | 1.39 | 54.93 | 65.06 | 2.28 |
| Traits a | Models | * | * | * | (SE) * |
|---|---|---|---|---|---|
| AGE | BLUP | 68.667 | 66.879 | 135.546 | 0.507 (0.0454) |
| ssGBLUP | 44.932 | 85.981 | 130.913 | 0.343 (0.0314) | |
| ADG | BLUP | 926.290 | 895.570 | 1821.860 | 0.508 (0.0453) |
| ssGBLUP | 581.4 | 1166.3 | 1747.7 | 0.333 (0.0308) | |
| BF | BLUP | 1.516 | 1.445 | 2.961 | 0.512 (0.0449) |
| ssGBLUP | 0.877 | 1.903 | 2.780 | 0.315 (0.0289) | |
| LMP | BLUP | 1.142 | 0.918 | 2.060 | 0.554 (0.0444) |
| ssGBLUP | 0.639 | 1.283 | 1.922 | 0.332 (0.0289) |
| Traits a | Chr b | Position (Mb) | nSNPs | gVar (%) c | Candidate Genes |
|---|---|---|---|---|---|
| AGE | 4 | 4.38–5.98 | 43 | 3.07 | FAM135B |
| 4 | 6.75–8.35 | 43 | 1.84 | ZFAT | |
| 14 | 1.63–3.23 | 22 | 1.57 | NFIL3, ROR2 | |
| ADG | 4 | 4.38–5.98 | 43 | 2.56 | FAM135B |
| 2 | 130.75–132.35 | 20 | 1.91 | SLC27A6 | |
| 2 | 149.94–151.54 | 29 | 1.49 | ADRB2 | |
| BF | 7 | 29.34–30.94 | 26 | 2.97 | DAXX, ITPR3, IP6K3, PACSIN1 |
| 3 | 117.76–119.36 | 19 | 1.94 | SDC1 | |
| 10 | 55.95–57.55 | 29 | 1.85 | NRP1 | |
| LMP | 2 | 8.11–9.71 | 26 | 2.68 | NAA40, LGALS12 |
| 3 | 117.76–119.36 | 39 | 2.08 | SDC1 | |
| 10 | 38.67–40.27 | 15 | 2.00 | MOB3B, RAB18, MPP7 |
| Term a | Count | p-Value | Genes |
|---|---|---|---|
| GO:0003727—single-stranded RNA binding | 4 | 0.004495 | SNRPC, NXF1, JMJD6, POLR2G |
| GO:0032435—negative regulation of proteasomal ubiquitin-dependent protein catabolic process | 3 | 0.020544 | WAC, UBXN1, SDCBP |
| GO:0002924—negative regulation of humoral immune response mediated by circulating immunoglobulin | 2 | 0.029686 | PTPN6, FOXJ1 |
| GO:0030335—positive regulation of cell migration | 5 | 0.031742 | ROR2, SEMA4D, CSF1R, SDCBP, SPHK1 |
| GO:0008076—voltage-gated potassium channel complex | 4 | 0.035714 | KCNC1, KCNJ11, KCNJ2, ABCC8 |
| GO:0005783—endoplasmic reticulum | 11 | 0.038199 | GPC2, CREB3L1, VWF, P3H3, BRINP1, ATL3, PLAAT3, EEF1G, SRP68, CLDN14, GANAB |
| GO:1904504—positive regulation of lipophagy | 2 | 0.044199 | ADRB2, SPTLC1 |
| GO:0032651—regulation of interleukin-1 β production | 2 | 0.044199 | S1PR3, SPHK1 |
| GO:0030501—positive regulation of bone mineralization | 3 | 0.049487 | ADRB2, OSR1, FBN2 |
| GO:0010107—potassium ion import | 3 | 0.049487 | KCNJ11, KCNJ16, KCNJ2 |
| ssc04742—taste transduction | 5 | 0.000381 | TAS1R1, GRM4, ITPR3, GNB3, SCNN1A |
| ssc04911—insulin secretion | 5 | 0.019468 | CREB3L1, KCNJ11, CAMK2A, ITPR3, ABCC8 |
| ssc04725—cholinergic synapse | 5 | 0.045538 | CREB3L1, CAMK2A, ITPR3, GNB3, KCNJ2 |
| ssc03320—PPAR signaling pathway | 4 | 0.047474 | ACOX1, SLC27A6, PLTP, CYP7A1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, D.; Zhuang, Z.; Ding, R.; Qiu, Y.; Zhou, S.; Wu, J.; Xu, C.; Hong, L.; Huang, S.; Zheng, E.; et al. Weighted Single-Step GWAS Identified Candidate Genes Associated with Growth Traits in a Duroc Pig Population. Genes 2021, 12, 117. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010117
Ruan D, Zhuang Z, Ding R, Qiu Y, Zhou S, Wu J, Xu C, Hong L, Huang S, Zheng E, et al. Weighted Single-Step GWAS Identified Candidate Genes Associated with Growth Traits in a Duroc Pig Population. Genes. 2021; 12(1):117. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010117
Chicago/Turabian StyleRuan, Donglin, Zhanwei Zhuang, Rongrong Ding, Yibin Qiu, Shenping Zhou, Jie Wu, Cineng Xu, Linjun Hong, Sixiu Huang, Enqin Zheng, and et al. 2021. "Weighted Single-Step GWAS Identified Candidate Genes Associated with Growth Traits in a Duroc Pig Population" Genes 12, no. 1: 117. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010117