Mapping the Transcriptional and Fitness Landscapes of a Pathogenic E. coli Strain: The Effects of Organic Acid Stress under Aerobic and Anaerobic Conditions


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Growth Conditions
2.2. Construction of TraDIS Library
2.3. RNA Extraction, Sequencing, and Data Analysis
2.4. TraDIS Methods
2.5. Additional Bioinformatics Analysis
3. Results
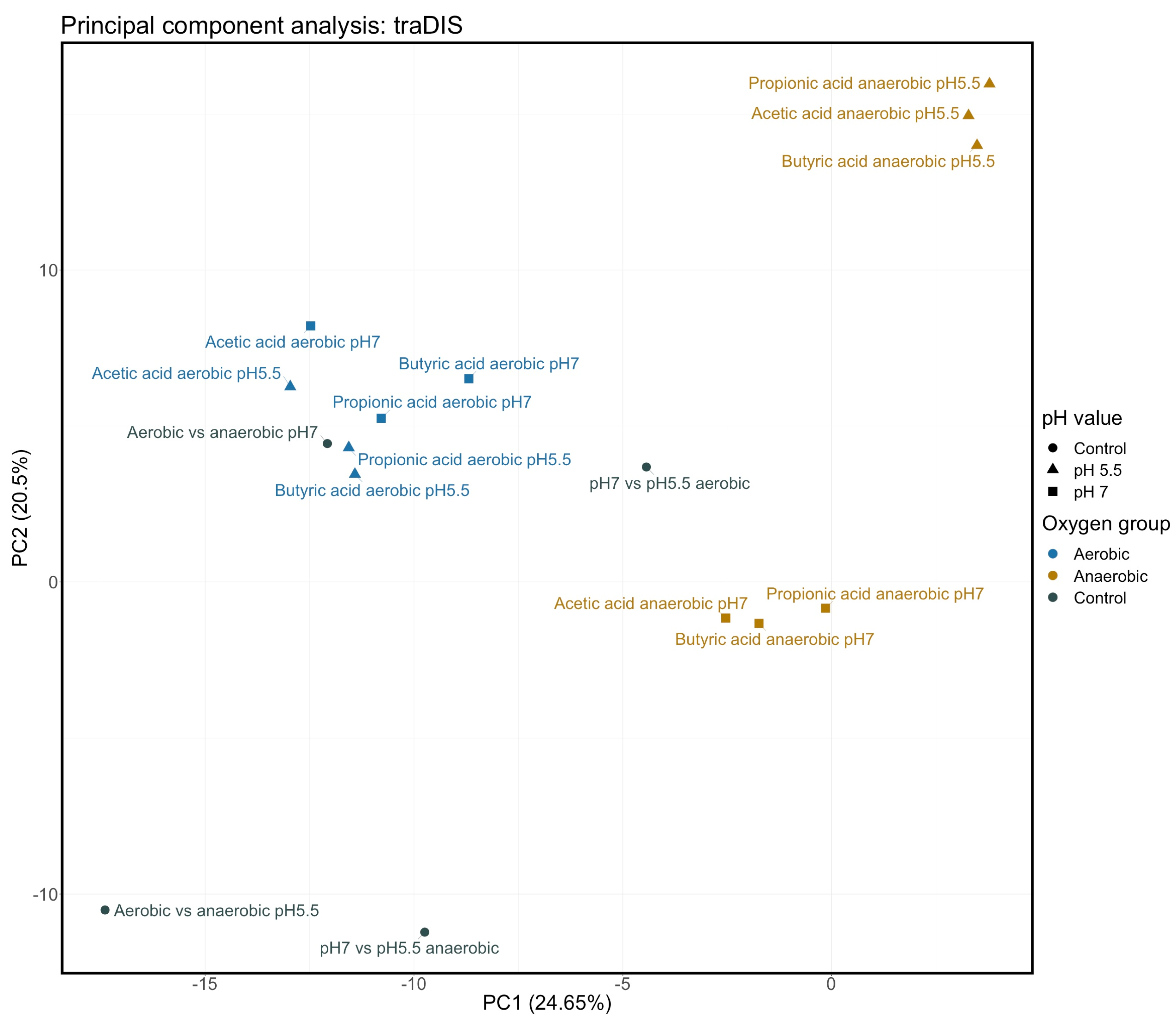
3.1. Determination of Stress Conditions
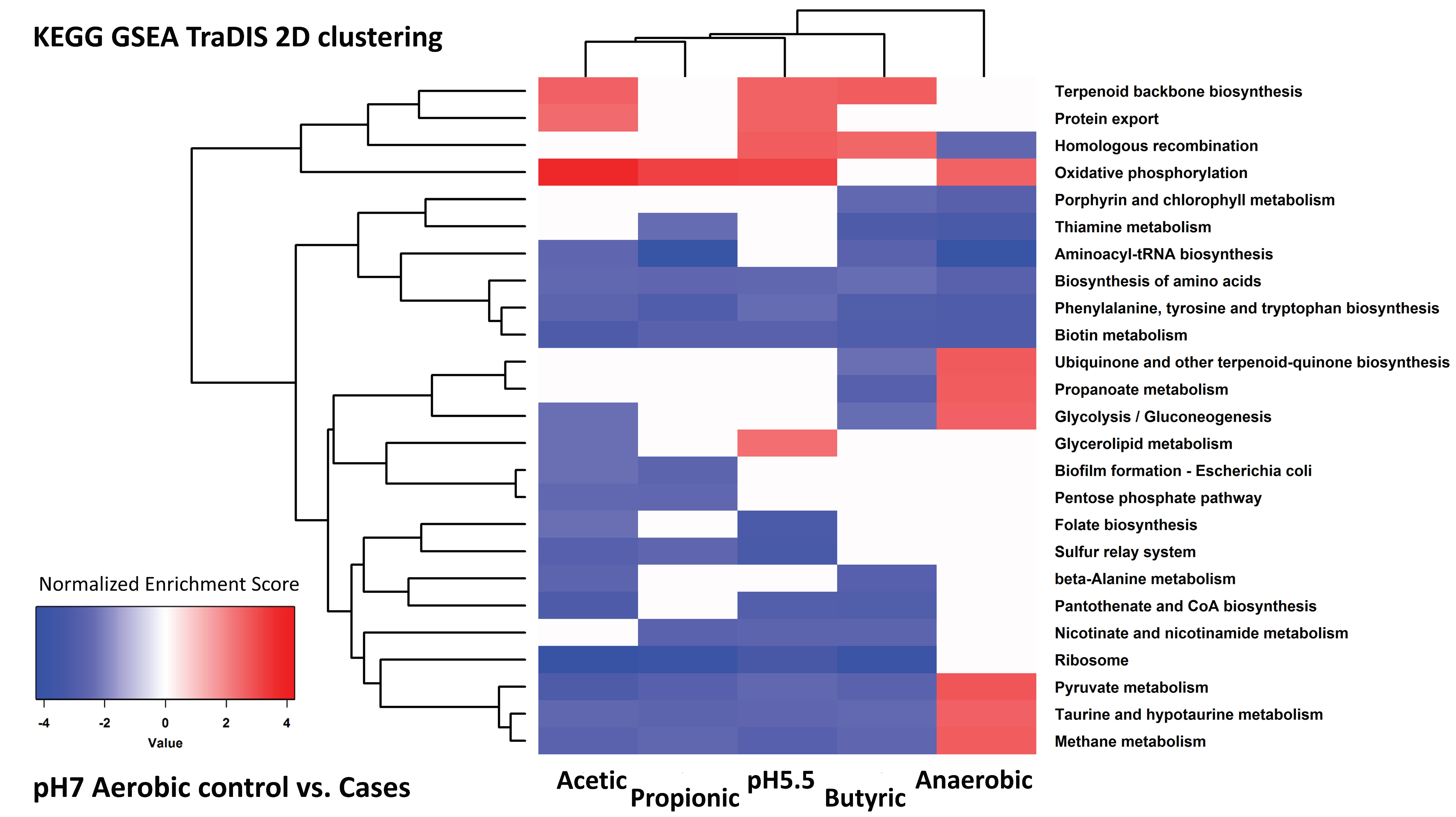
3.2. Analysis of Gene Expression
3.3. Analysis of Contributions of Genes and Pathways to Strain Fitness
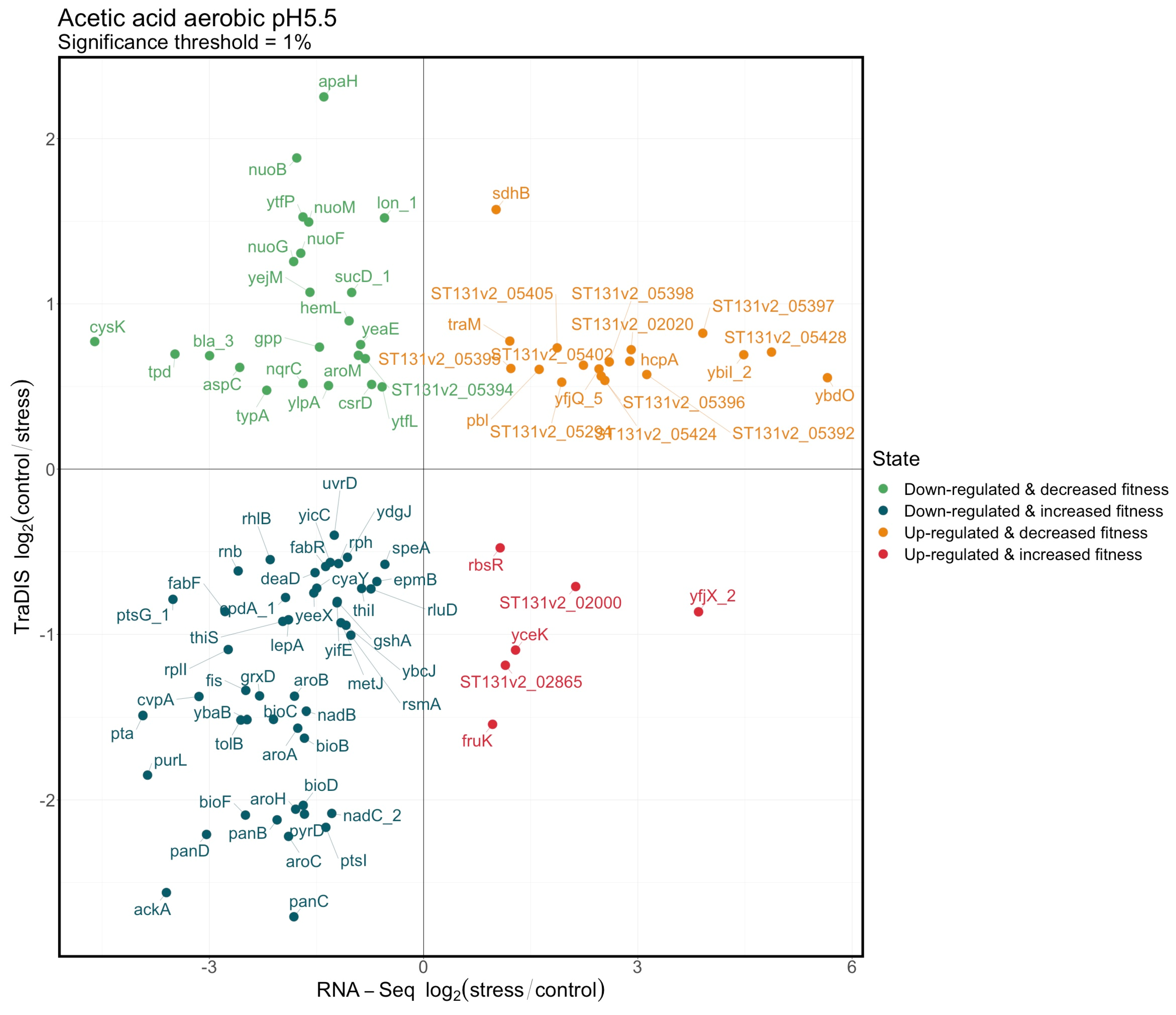
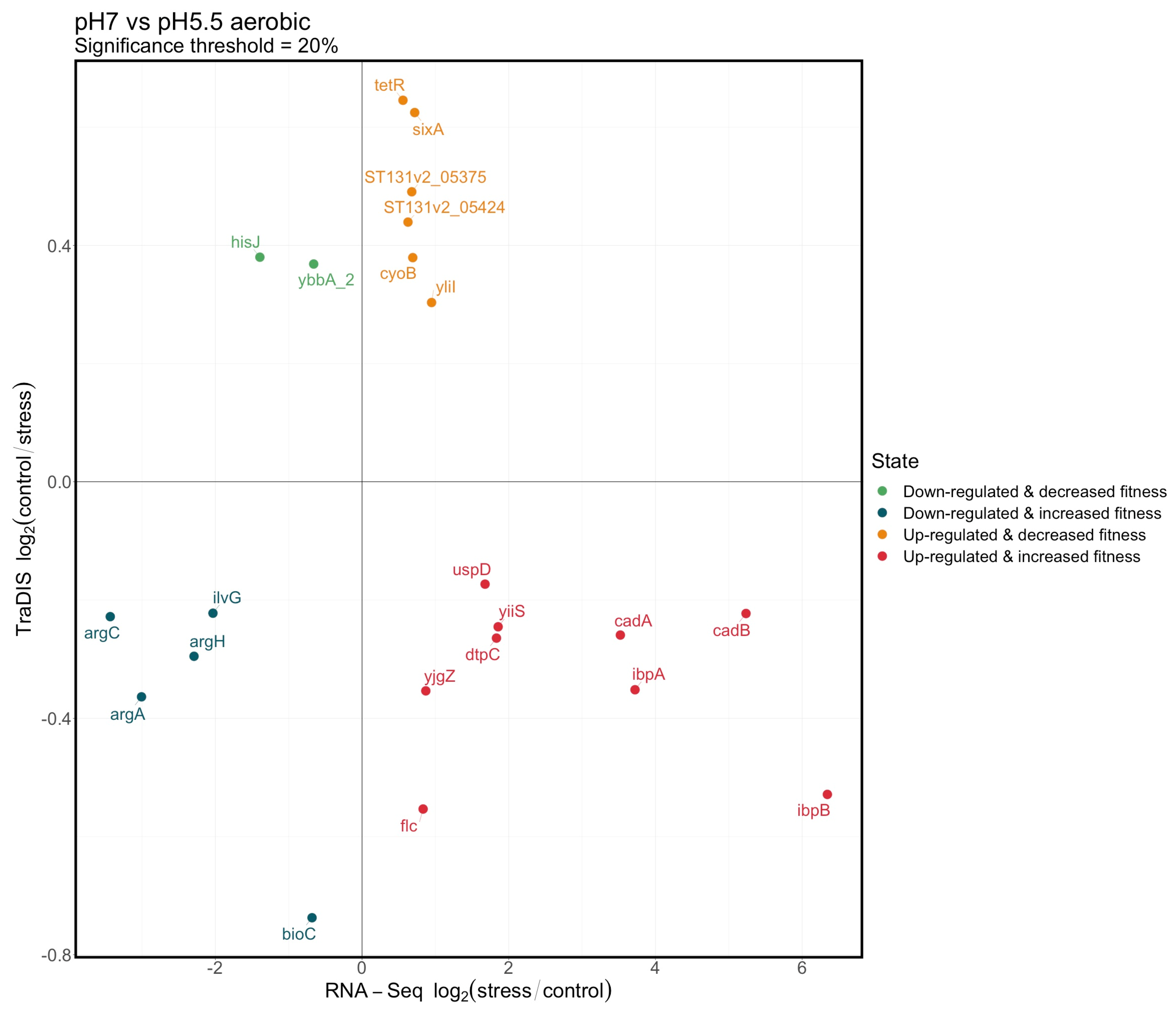
3.4. Integration of the RNAseq and TraDIS Analyses to Distinguish Adaptive and Adverse Responses
3.5. Non-Core Genes Show Significantly Enriched Correlations between Expression Level and Contribution to Strain Fitness
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, E.J.; A Lerman, J.; Chang, R.L.; Hyduke, D.R.; Palsson, B.O. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013, 9, 693. [Google Scholar] [CrossRef] [PubMed]
- Carrera, J.; Estrela, R.; Luo, J.; Rai, N.; Tsoukalas, A.; Tagkopoulos, I. An integrative, multi-scale, genome-wide model reveals the phenotypic landscape of E scherichia coli. Mol. Syst. Biol. 2014, 10, 735. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Rai, N.; Zorraquino, V.; Tagkopoulos, I. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 2016, 7, 13090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galardini, M.; Koumoutsi, A.; Herrera-Dominguez, L.; Varela, J.A.C.; Telzerow, A.; Wagih, O.; Wartel, M.; Clermont, O.; Denamur, E.; Typas, A.; et al. Phenotype inference in an Escherichia coli strain panel. eLife 2017, 6, e31035. [Google Scholar] [CrossRef]
- Price, M.N.; Wetmore, K.M.; Waters, R.J.; Callaghan, M.; Ray, J.; Liu, H.; Kuehl, J.V.; Melnyk, R.A.; Lamson, J.S.; Suh, Y.; et al. Mutant phenotypes for thousands of bacterial genes of unknown function. Nat. Cell Biol. 2018, 557, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.; A King, Z.; Sastry, A.; Palsson, B.O. The y-ome defines the 35% ofEscherichia coligenes that lack experimental evidence of function. Nucleic Acids Res. 2019, 47, 2446–2454. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Gunawardena, J. Models in Systems Biology: The Parameter Problem and the Meanings of Robustness. In Elements of Computational Systems Biology; Lodhi, H.M., Muggleton, S.H., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2010; pp. 19–47. [Google Scholar]
- Han, M.-J.; Lee, S.Y. The Escherichia coli Proteome: Past, Present, and Future Prospects. Microbiol. Mol. Biol. Rev. 2006, 70, 362–439. [Google Scholar] [CrossRef] [Green Version]
- Abreu, R.D.S.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Q.; Zhang, B. Leveraging the complementary nature of RNA-Seq and shotgun proteomics data. Proteomics 2014, 14, 2676–2687. [Google Scholar] [CrossRef] [PubMed]
- Wiser, M.J.; Lenski, R.E. A Comparison of Methods to Measure Fitness in Escherichia coli. PLoS ONE 2015, 10, e0126210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.L.; Mekalanos, J.J.; Holden, D.W. In Vivo Genetic Analysis of Bacterial Virulence. Annu. Rev. Microbiol. 1999, 53, 129–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, F. Transposon-Based Strategies for Microbial Functional Genomics and Proteomics. Annu. Rev. Genet. 2003, 37, 3–29. [Google Scholar] [CrossRef]
- Cummins, J.; Gahan, C.G.M. Signature tagged mutagenesis in the functional genetic analysis of gastrointestinal pathogens. Gut Microbes 2012, 3, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Van Opijnen, T.; Camilli, A. Transposon insertion sequencing: A new tool for systems-level analysis of microorganisms. Nat. Rev. Genet. 2013, 11, 435–442. [Google Scholar] [CrossRef]
- Cain, A.K.; Barquist, L.; Goodman, A.L.; Paulsen, I.T.; Parkhill, J.; Van Opijnen, T. A decade of advances in transposon-insertion sequencing. Nat. Rev. Genet. 2020, 21, 526–540. [Google Scholar] [CrossRef]
- Nicolas-Chanoine, M.-H.; Bertrand, X.; Madec, J.-Y. Escherichia coli ST131, an Intriguing Clonal Group. Clin. Microbiol. Rev. 2014, 27, 543–574. [Google Scholar] [CrossRef] [Green Version]
- Russo, T.A.; Stapleton, A.; Wenderoth, S.; Hooton, T.M.; Stamm, W.E. Chromosomal Restriction Fragment Length Polymorphism Analysis of Escherichia coli Strains Causing Recurrent Urinary Tract Infections in Young Women. J. Infect. Dis. 1995, 172, 440–445. [Google Scholar] [CrossRef]
- Forde, B.M.; Roberts, L.W.; Phan, M.-D.; Peters, K.M.; Fleming, B.A.; Russell, C.W.; Lenherr, S.M.; Myers, J.B.; Barker, A.P.; Fisher, M.A.; et al. Population dynamics of an Escherichia coli ST131 lineage during recurrent urinary tract infection. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Berry, E.D.; Cutter, C.N. Effects of Acid Adaptation of Escherichia coli O157:H7 on Efficacy of Acetic Acid Spray Washes To Decontaminate Beef Carcass Tissue. Appl. Environ. Microbiol. 2000, 66, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Richard, H.T.; Foster, J.W. Acid Resistance in Escherichia coli. Adv. Clin. Chem. 2003, 52, 167–186. [Google Scholar] [CrossRef]
- Foster, J.W. Escherichia coli acid resistance: Tales of an amateur acidophile. Nat. Rev. Genet. 2004, 2, 898–907. [Google Scholar] [CrossRef]
- Kanjee, U.; Houry, W.A. Mechanisms of Acid Resistance inEscherichia coli. Annu. Rev. Microbiol. 2013, 67, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biase, D.; Lund, P.A. The Escherichia coli Acid Stress Response and Its Significance for Pathogenesis. Adv. Appl. Microbiol. 2015, 92, 49–88. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Smith, M.P.; Chapin, K.C.; Baik, H.S.; Bennett, G.N.; Foster, J.W. Mechanisms of acid resistance in enterohemorrhagic Escherichia coli. Appl. Environ. Microbiol. 1996, 62, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Large, T.M.; Walk, S.T.; Whittam, T.S. Variation in Acid Resistance among Shiga Toxin-Producing Clones of Pathogenic Escherichia coli. Appl. Environ. Microbiol. 2005, 71, 2493–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergholz, T.M.; Whittam, T.S. Variation in acid resistance among enterohaemorrhagic Escherichia coli in a simulated gastric environment. J. Appl. Microbiol. 2007, 102, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.J.; McLaggan, D.; Davidson, I.; O’Byrne, C.; Booth, I.R. Perturbation of Anion Balance during Inhibition of Growth of Escherichia coli by Weak Acids. J. Bacteriol. 1998, 180, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Slonczewski, J.L.; Fujisawa, M.; Dopson, M.; Krulwich, T.A. Cytoplasmic pH Measurement and Homeostasis in Bacteria and Archaea. Adv. Microb. Physiol. 2009, 55, 1–317. [Google Scholar] [CrossRef]
- Blankenhorn, D.; Phillips, J.; Slonczewski, J.L. Acid- and Base-Induced Proteins during Aerobic and Anaerobic Growth of Escherichia coli Revealed by Two-Dimensional Gel Electrophoresis. J. Bacteriol. 1999, 181, 2209–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, K.; Riggins, D.P.; Eldahan, K.C.; Kitko, R.D.; Slonczewski, J.L. Hydrogenase-3 Contributes to Anaerobic Acid Resistance of Escherichia coli. PLoS ONE 2010, 5, e10132. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Shan, Y.; Pan, Q.; Gao, X.; Yan, A. Anaerobic expression of the gadE-mdtEF multidrug efflux operon is primarily regulated by the two-component system ArcBA through antagonizing the H-NS mediated repression. Front. Microbiol. 2013, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Woodford, N.; Carattoli, A.; Karisik, E.; Underwood, A.; Ellington, M.J.; Livermore, D.M. Complete Nucleotide Sequences of Plasmids pEK204, pEK499, and pEK516, Encoding CTX-M Enzymes in Three Major Escherichia coli Lineages from the United Kingdom, All Belonging to the International O25:H4-ST131 Clone. Antimicrob. Agents Chemother. 2009, 53, 4472–4482. [Google Scholar] [CrossRef] [Green Version]
- Burton, N.A.; Johnson, M.D.; Antczak, P.; Robinson, A.; Lund, P.A. Novel Aspects of the Acid Response Network of E. coli K-12 Are Revealed by a Study of Transcriptional Dynamics. J. Mol. Biol. 2010, 401, 726–742. [Google Scholar] [CrossRef]
- Yasir, M.; Turner, A.K.; Bastkowski, S.; Baker, D.; Page, A.J.; Telatin, A.; Phan, M.-D.; Monahan, L.; Savva, G.M.; Darling, A.; et al. TraDIS-Xpress: A high-resolution whole-genome assay identifies novel mechanisms of triclosan action and resistance. Genome Res. 2020, 30, 239–249. [Google Scholar] [CrossRef]
- Stincone, A.; Daudi, N.; Rahman, A.S.; Antczak, P.; Henderson, I.; Cole, J.; Johnson, M.D.; Lund, P.; Falciani, F. A systems biology approach sheds new light on Escherichia coli acid resistance. Nucleic Acids Res. 2011, 39, 7512–7528. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Goodall, E.C.A.; Robinson, A.; Johnston, I.G.; Jabbari, S.; Turner, K.A.; Cunningham, A.F.; Lund, P.A.; Cole, J.A.; Henderson, I.R. The Essential Genome of Escherichia coli K-12. MBio 2018, 9, 02096-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.L.; Burghout, P.; Bootsma, H.J.; Hermans, P.W.M.; Van Hijum, S.A.F.T. ESSENTIALS: Software for Rapid Analysis of High Throughput Transposon Insertion Sequencing Data. PLoS ONE 2012, 7, e43012. [Google Scholar] [CrossRef]
- R Core Team, R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Italy, 2018. [Google Scholar]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. gplots: Various R programming tools for plotting data. R Package Version 2019, 2, 1. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Ihssen, J.; Egli, T. Specific growth rate and not cell density controls the general stress response in Escherichia coli. Microbiology 2004, 150, 1637–1648. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.H. Short chain fatty acids in the human colon. Gut 1981, 22, 763–779. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Charlier, D.; Bervoets, I. Regulation of arginine biosynthesis, catabolism and transport in Escherichia coli. Amino Acids 2019, 51, 1103–1127. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Dodson, K.W.; Kalas, V.; Chen, S.L.; Hultgren, S.J. Adaptation of Arginine Synthesis among Uropathogenic Branches of the Escherichia coli Phylogeny Reveals Adjustment to the Urinary Tract Habitat. MBio 2020, 11, 02318–02320. [Google Scholar] [CrossRef]
- Richmond, C.S.; Glasner, J.D.; Mau, R.; Jin, H.; Blattner, F.R. Genome-wide expression profiling in Escherichia coli K-12. Nucleic Acids Res. 1999, 27, 3821–3835. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.G.; Baneyx, F. Roles of the Escherichia coli Small Heat Shock Proteins IbpA and IbpB in Thermal Stress Management: Comparison with ClpA, ClpB, and HtpG In Vivo. J. Bacteriol. 1998, 180, 5165–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birrell, G.W.; Brown, J.M.; Wu, H.I.; Giaever, G.; Chu, A.M.; Davis, R.W.; Brown, J.M. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc. Natl. Acad. Sci. USA 2002, 99, 8778–8783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Deutschbauer, A.M.; Skerker, J.M.; Wetmore, K.M.; Ruths, T.; Mar, J.S.; Kuehl, J.V.; Shao, W.; Arkin, A.P. Indirect and suboptimal control of gene expression is widespread in bacteria. Mol. Syst. Biol. 2013, 9, 660. [Google Scholar] [CrossRef]
- Keren, L.; Hausser, J.; Lotan-Pompan, M.; Slutskin, I.V.; Alisar, H.; Kaminski, S.; Weinberger, A.; Alon, U.; Milo, R.; Segal, E. Massively Parallel Interrogation of the Effects of Gene Expression Levels on Fitness. Cell 2016, 166, 1282–1294.e18. [Google Scholar] [CrossRef]
- Phan, M.-D.; Peters, K.M.; Sarkar, S.; Lukowski, S.W.; Allsopp, L.P.; Moriel, D.G.; Achard, M.E.S.; Totsika, M.; Marshall, V.M.; Upton, M.; et al. The Serum Resistome of a Globally Disseminated Multidrug Resistant Uropathogenic Escherichia coli Clone. PLoS Genet. 2013, 9, e1003834. [Google Scholar] [CrossRef] [Green Version]
- Khatiwara, A.; Jiang, T.; Sung, S.-S.; Dawoud, T.; Kim, J.N.; Bhattacharya, D.; Kim, H.-B.; Ricke, S.C.; Kwon, Y.M. Genome Scanning for Conditionally Essential Genes in Salmonella enterica Serotype Typhimurium. Appl. Environ. Microbiol. 2012, 78, 3098–3107. [Google Scholar] [CrossRef] [Green Version]
- Langridge, G.C.; Phan, M.-D.; Turner, D.J.; Perkins, T.T.; Parts, L.; Haase, J.; Charles, I.; Maskell, D.J.; Peters, S.E.; Dougan, G.; et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009, 19, 2308–2316. [Google Scholar] [CrossRef] [Green Version]
- Van Opijnen, T.; Camilli, A. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 2012, 22, 2541–2551. [Google Scholar] [CrossRef] [Green Version]
- Deutschbauer, A.; Price, M.N.; Wetmore, K.M.; Shao, W.; Baumohl, J.K.; Xu, Z.; Nguyen, M.; Tamse, R.; Davis, R.W.; Arkin, A.P. Evidence-Based Annotation of Gene Function in Shewanella oneidensis MR-1 Using Genome-Wide Fitness Profiling across 121 Conditions. PLoS Genet. 2011, 7, e1002385. [Google Scholar] [CrossRef] [Green Version]
- Gaubig, L.C.; Waldminghaus, T.; Narberhaus, F. Multiple layers of control govern expression of the Escherichia coli ibpAB heat-shock operon. Microbiology 2011, 157, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.L. The Lysine Decarboxylase CadA Protects Escherichia coli Starved of Phosphate against Fermentation Acids. J. Bacteriol. 2007, 189, 2249–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmond, C.V.; Kroll, R.G.; Booth, I.R. The Effect of Food Preservatives on pH Homeostasis in Escherichia coli. Microbiology 1984, 130, 2845–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trcek, J.; Mira, N.P.; Jarboe, L.R. Adaptation and tolerance of bacteria against acetic acid. Appl. Microbiol. Biotechnol. 2015, 99, 6215–6229. [Google Scholar] [CrossRef]
- Hirshfield, I.N.; Terzulli, S.; O’Byrne, C. Weak Organic Acids: A Panoply of Effects on Bacteria. Sci. Prog. 2003, 86, 245–270. [Google Scholar] [CrossRef]
- Pinhal, S.; Ropers, D.; Geiselmann, J.; De Jong, H. Acetate Metabolism and the Inhibition of Bacterial Growth by Acetate. J. Bacteriol. 2019, 201, 00147-19. [Google Scholar] [CrossRef] [Green Version]
- Bjarnsholt, T.; Alhede, M.; Jensen, P.Ø.; Nielsen, A.K.; Johansen, H.K.; Homøe, P.; Høiby, N.; Givskov, M.C.; Kirketerp-Møller, K. Antibiofilm Properties of Acetic Acid. Adv. Wound Care 2015, 4, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Halstead, F.D.; Rauf, M.; Moiemen, N.; Bamford, A.; Wearn, C.M.; Fraise, A.P.; Lund, P.A.; Oppenheim, B.A.; Webber, M.A. The Antibacterial Activity of Acetic Acid against Biofilm-Producing Pathogens of Relevance to Burns Patients. PLoS ONE 2015, 10, e0136190. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.P.; Cronan, J.E. Two-Carbon Compounds and Fatty Acids as Carbon Sources. EcoSal Plus 2005, 1. [Google Scholar] [CrossRef]
- Wolfe, A.J. The Acetate Switch. Microbiol. Mol. Biol. Rev. 2005, 69, 12–50. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; O’Riordan, M.X.D. Regulation of Bacterial Pathogenesis by Intestinal Short-Chain Fatty Acids. Adv. Appl. Microbiol. 2013, 85, 93–118. [Google Scholar] [PubMed] [Green Version]
- Ormsby, M.J.; Johnson, S.A.; Carpena, N.; Meikle, L.M.; Goldstone, R.J.; McIntosh, A.; Wessel, H.M.; Hulme, H.E.; McConnachie, C.C.; Connolly, J.P.; et al. Propionic Acid Promotes the Virulent Phenotype of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Rep. 2020, 30, 2297–2305.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, C.; Maurer, L.M.; Oyelakin, N.E.; Yoncheva, Y.N.; Maurer, R.; Slonczewski, J.L. Acetate and Formate Stress: Opposite Responses in the Proteome of Escherichia coli. J. Bacteriol. 2001, 183, 6466–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.; Lucchini, S.; Hinton, J.C.D.; Gobius, K. Transcriptomic Analysis of Escherichia coli O157:H7 and K-12 Cultures Exposed to Inorganic and Organic Acids in Stationary Phase Reveals Acidulant- and Strain-Specific Acid Tolerance Responses. Appl. Environ. Microbiol. 2010, 76, 6514–6528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobe, T.; Nakanishi, N.; Sugimoto, N. Activation of Motility by Sensing Short-Chain Fatty Acids via Two Steps in a Flagellar Gene Regulatory Cascade in EnterohemorrhagicEscherichia coli. Infect. Immun. 2011, 79, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Polen, T.; Rittmann, D.; Wendisch, V.F.; Sahm, H. DNA Microarray Analyses of the Long-Term Adaptive Response of Escherichia coli to Acetate and Propionate. Appl. Environ. Microbiol. 2003, 69, 1759–1774. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bushell, F.; Herbert, J.M.J.; Sannasiddappa, T.H.; Warren, D.; Turner, A.K.; Falciani, F.; Lund, P.A. Mapping the Transcriptional and Fitness Landscapes of a Pathogenic E. coli Strain: The Effects of Organic Acid Stress under Aerobic and Anaerobic Conditions. Genes 2021, 12, 53. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010053
Bushell F, Herbert JMJ, Sannasiddappa TH, Warren D, Turner AK, Falciani F, Lund PA. Mapping the Transcriptional and Fitness Landscapes of a Pathogenic E. coli Strain: The Effects of Organic Acid Stress under Aerobic and Anaerobic Conditions. Genes. 2021; 12(1):53. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010053
Chicago/Turabian StyleBushell, Francesca, John M. J. Herbert, Thippeswamy H. Sannasiddappa, Daniel Warren, A. Keith Turner, Francesco Falciani, and Peter A. Lund. 2021. "Mapping the Transcriptional and Fitness Landscapes of a Pathogenic E. coli Strain: The Effects of Organic Acid Stress under Aerobic and Anaerobic Conditions" Genes 12, no. 1: 53. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010053