
MYO5A Frameshift Variant in a Miniature Dachshund with Coat Color Dilution and Neurological Defects Resembling Human Griscelli Syndrome Type 1

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Examination
2.2. Necropsy, Histopathology, and Immunohistochemistry
2.3. Control Samples for Genetic Analyses
2.4. DNA Extraction
2.5. Whole-Genome Sequencing
2.6. Variant Calling
2.7. Gene Analysis
2.8. PCR and Sanger Sequencing
3. Results
3.1. Clinical Examination
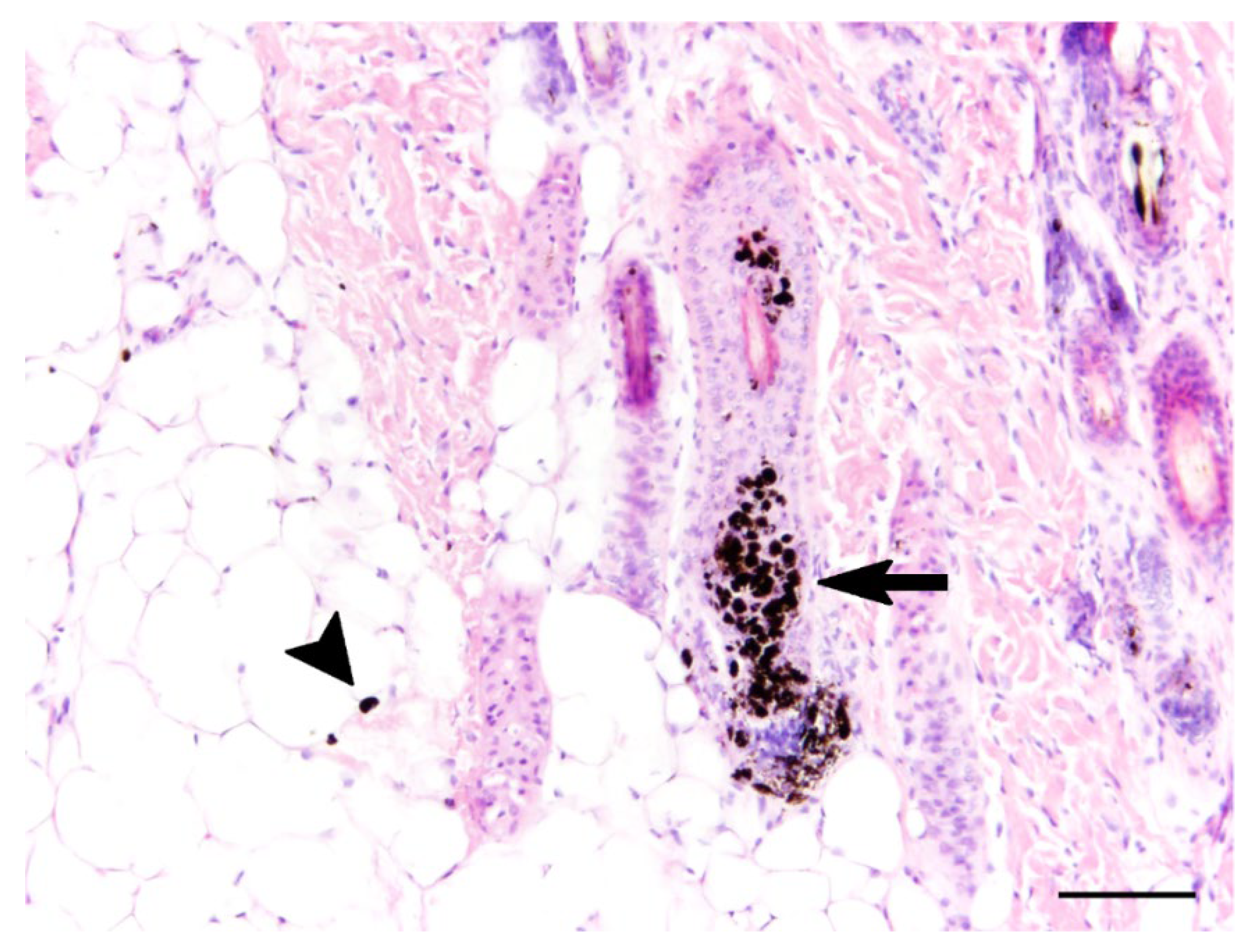
3.2. Gross, Histopathological, and Immunohistochemical Findings
3.3. Genetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griscelli, C.; Durandy, A.; Guy-Grand, D.; Daguillard, F.; Herzog, C.; Prunieras, M. A syndrome associating partial albinism and immunodeficiency. Am. J. Med. 1978, 65, 691–702. [Google Scholar] [CrossRef]
- Pastural, E.; Barrat, F.J.; Dufourcq-Lagelouse, R.; Certain, S.; Sanal, O.; Jabado, N.; Seger, R.; Griscelli, C.; Fischer, A.; Basile, G.; et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the Myosin-Va gene. Nat. Genet. 1997, 16, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Menasche, G.; Fischer, A.; de Saint Basile, G. Griscelli Syndrome Types 1 and 2. Am. J. Hum. Genet. 2002, 71, 1237–1238. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.R.; Walker, L.J.; Pullaperuma, S.; Cooper, B.; Brueton, L.A.; de Saint Basile, G.; Suri, M.; Brady, A.F. Griscelli syndrome type 1: A report of two cases and review of the literature. Clin. Dysmorphol. 2009, 18, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Menasche, G.; Pastural, E.; Feldmann, J.; Certain, S.; Ersoy, F.; Dupuis, S.; Wulffraat, N.; Bianchi, D.; Fischer, A.; Le Deist, F.; et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 2000, 25, 173–176. [Google Scholar] [CrossRef]
- Menasche, G.; Ho, C.H.; Sanal, O.; Feldmann, J.; Tezcan, I.; Ersoy, F.; Houdusse, A.; Fischer, A.; Basile, G.D.S. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J. Clin. Investig. 2003, 112, 450–456. [Google Scholar] [CrossRef]
- Wu, X.; Bowers, B.; Rao, K.; Wei, Q.; Hammer, J.A. Visualization of Melanosome Dynamics within Wild-Type and Dilute Melanocytes Suggests a Paradigm for Myosin V Function In Vivo. J. Cell Biol. 1998, 143, 1899–1918. [Google Scholar] [CrossRef] [Green Version]
- Westbroek, W.; Lambert, J.; Naeyaert, J.M. The dilute locus and Griscelli syndrome: Gateways towards a better understanding of melanosome transport. Pigment Cell Res. 2001, 14, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Hammer, J.A. Melanosome transfer: It is best to give and receive. Curr. Opin. Cell Biol. 2014, 29, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Brenowitz, S.D.; Hammer, J.A. Myosin-Va transports the endoplasmic reticulum into the dendritic spines of Purkinje neurons. Nature 2010, 13, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Simpson, P.B.; Challiss, R.A.J.; Nahorski, S.R. Neuronal Ca2+ stores: Activation and function. Trends Neurosci. 1995, 18, 299–306. [Google Scholar] [CrossRef]
- Bollimuntha, S.; Pani, B.; Singh, B.B. Neurological and Motor Disorders: Neuronal Store-Operated Ca2+ Signaling: An Overview and Its Function. In Store-Operated Ca2+ Entry (SOCE) Pathways; Springer: Berlin/Heidelberg, Germany, 2017; Volume 993, pp. 535–556. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.A.; Seperack, P.K.; Strobel, M.C.; Copeland, N.G.; Jenkins, N.A. Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature 1991, 349, 709–713. [Google Scholar] [CrossRef]
- Takagishi, Y.; Murata, Y. Myosin Va Mutation in Rats Is an Animal Model for the Human Hereditary Neurological Disease, Griscelli Syndrome Type 1. Ann. N. Y. Acad. Sci. 2006, 1086, 66–80. [Google Scholar] [CrossRef]
- Brooks, S.A.; Gabreski, N.; Miller, D.; Brisbin, A.; Brown, H.E.; Streeter, C.; Mezey, J.; Cook, D.; Antczak, U.F. Whole-Genome SNP Association in the Horse: Identification of a Deletion in Myosin Va Responsible for Lavender Foal Syndrome. PLoS Genet. 2010, 6, e1000909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Zhao, W.-M.; Tang, B.-X.; Wang, Y.-Q.; Wang, L.; Zhang, Z.; Yang, H.-C.; Liu, Y.-H.; Zhu, J.-W.; Irwin, D.M.; et al. DoGSD: The dog and wolf genome SNP database. Nucleic Acids Res. 2014, 43, D777–D783. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drögemüller, C.; Philipp, U.; Haase, B.; Günzel-Apel, A.-R.; Leeb, T. A Noncoding Melanophilin Gene (MLPH) SNP at the Splice Donor of Exon 1 Represents a Candidate Causal Mutation for Coat Color Dilution in Dogs. J. Hered. 2007, 98, 468–473. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.; Kehl, A.; Jagannathan, V.; Leeb, T. A novelMLPHvariant in dogs with coat colour dilution. Anim. Genet. 2018, 49, 94–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Buren, S.L.; Minor, K.M.; Grahn, R.A.; Mickelson, J.R.; Grahn, J.C.; Malvick, J.; Colangelo, J.R.; Mueller, E.; Kuehnlein, P.; Kehl, A. A Third MLPH Variant Causing Coat Color Dilution in Dogs. Genes 2020, 11, 639. [Google Scholar] [CrossRef]
- Chieffo, C.; Stalis, I.H.; Winkle, T.J.; Haskins, M.E.; Patterson, D.F. Cerebellar Purkinje’s Cell Degeneration and Coat Color Dilution in a Family of Rhodesian Ridgeback Dogs. J. Veter. Intern. Med. 1994, 8, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Welle, M.; Philipp, U.; Rufenacht, S.; Roosje, P.; Scharfenstein, M.; Schutz, E.; Brenig, B.; Linek, M.; Mecklenburg, L.; Grest, P.; et al. MLPH Genotype--Melanin Phenotype Correlation in Dilute Dogs. J. Hered. 2009, 100, S75–S79. [Google Scholar] [CrossRef] [Green Version]
- Perego, R.; Proverbio, D.; Roccabianca, P.; Spada, E. Color dilution alopecia in a blue Doberman pinscher crossbreed. Can. Vet. J. 2009, 50, 511–514. [Google Scholar] [PubMed]
- Li, J.-F.; Nebenführ, A. The Tail that Wags the Dog: The Globular Tail Domain Defines the Function of Myosin V/XI. Traffic 2008, 9, 290–298. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Homozygous Variants |
|---|---|
| All variants in the affected miniature Dachshund | 2,698,983 |
| Private variants | 1688 |
| Protein-changing 1 private variants | 12 |
| Protein-changing 1 private variants in MYO5A | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christen, M.; de le Roi, M.; Jagannathan, V.; Becker, K.; Leeb, T. MYO5A Frameshift Variant in a Miniature Dachshund with Coat Color Dilution and Neurological Defects Resembling Human Griscelli Syndrome Type 1. Genes 2021, 12, 1479. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101479
Christen M, de le Roi M, Jagannathan V, Becker K, Leeb T. MYO5A Frameshift Variant in a Miniature Dachshund with Coat Color Dilution and Neurological Defects Resembling Human Griscelli Syndrome Type 1. Genes. 2021; 12(10):1479. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101479
Chicago/Turabian StyleChristen, Matthias, Madeleine de le Roi, Vidhya Jagannathan, Kathrin Becker, and Tosso Leeb. 2021. "MYO5A Frameshift Variant in a Miniature Dachshund with Coat Color Dilution and Neurological Defects Resembling Human Griscelli Syndrome Type 1" Genes 12, no. 10: 1479. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101479