
EWAS of Monozygotic Twins Implicate a Role of mTOR Pathway in Pathogenesis of Tic Spectrum Disorder

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Individuals, Phenotyping and Zygosity Studies
2.2. DNA Methylation Profiling and Quality Control
2.3. Statistical Analyses
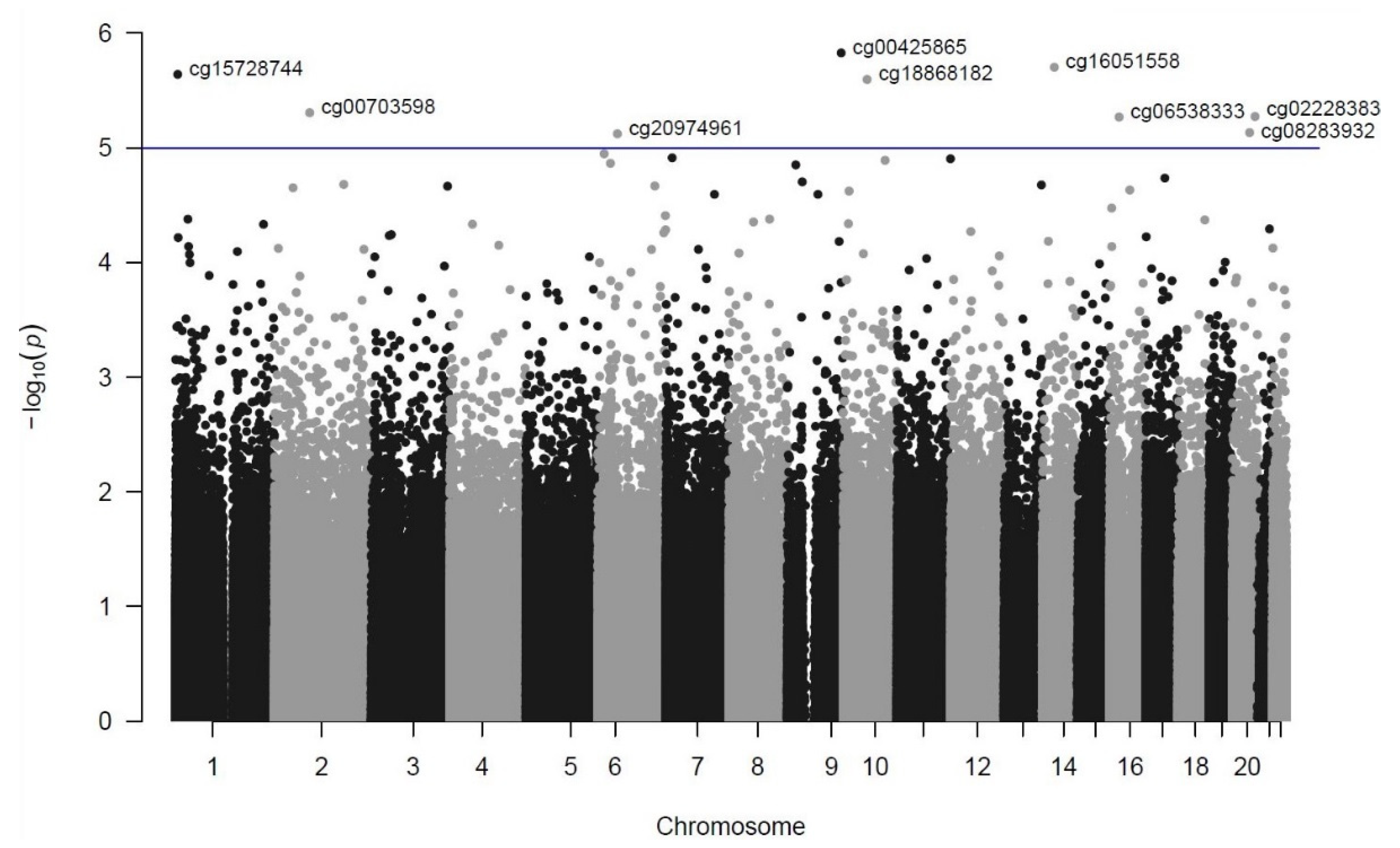
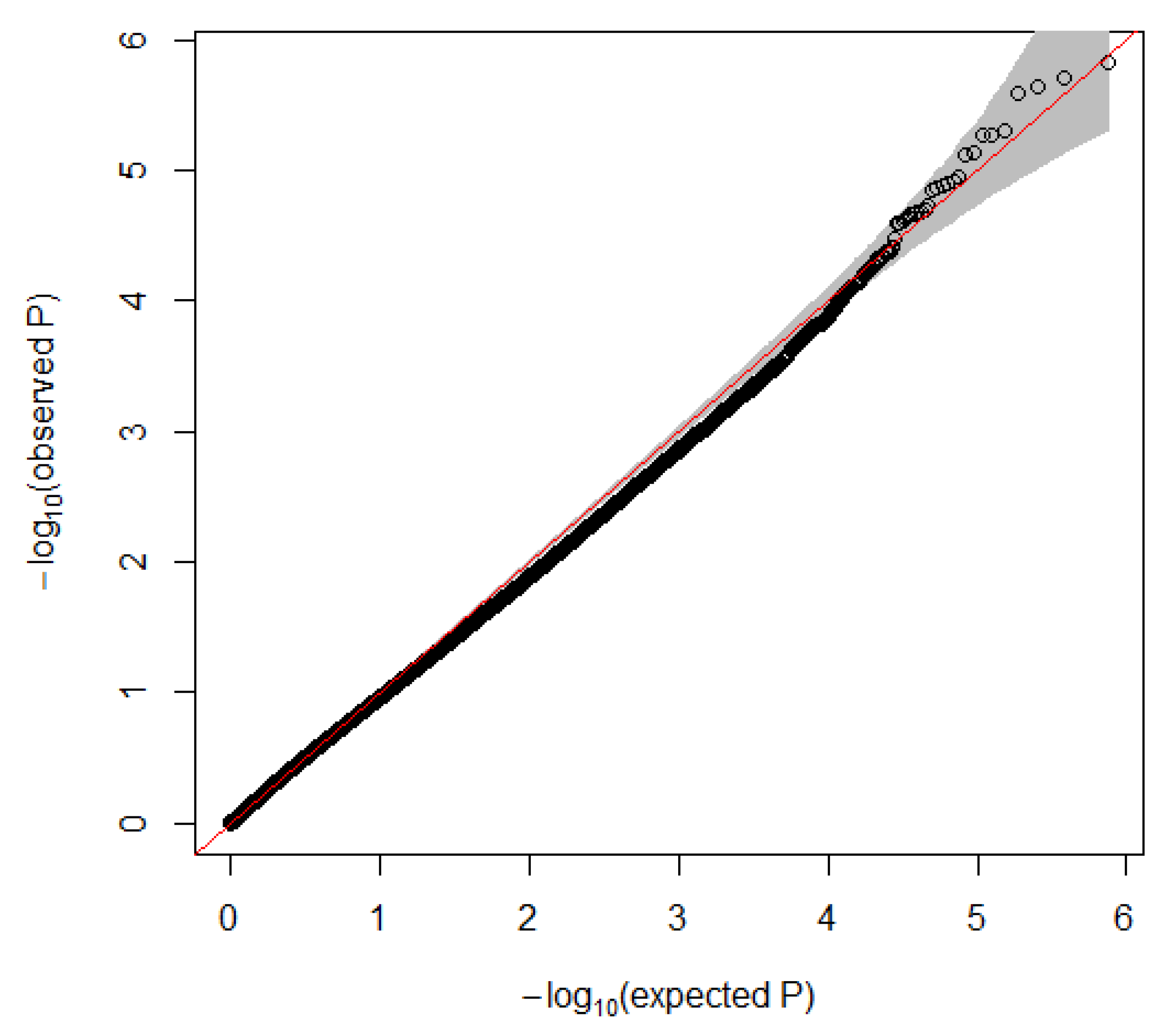
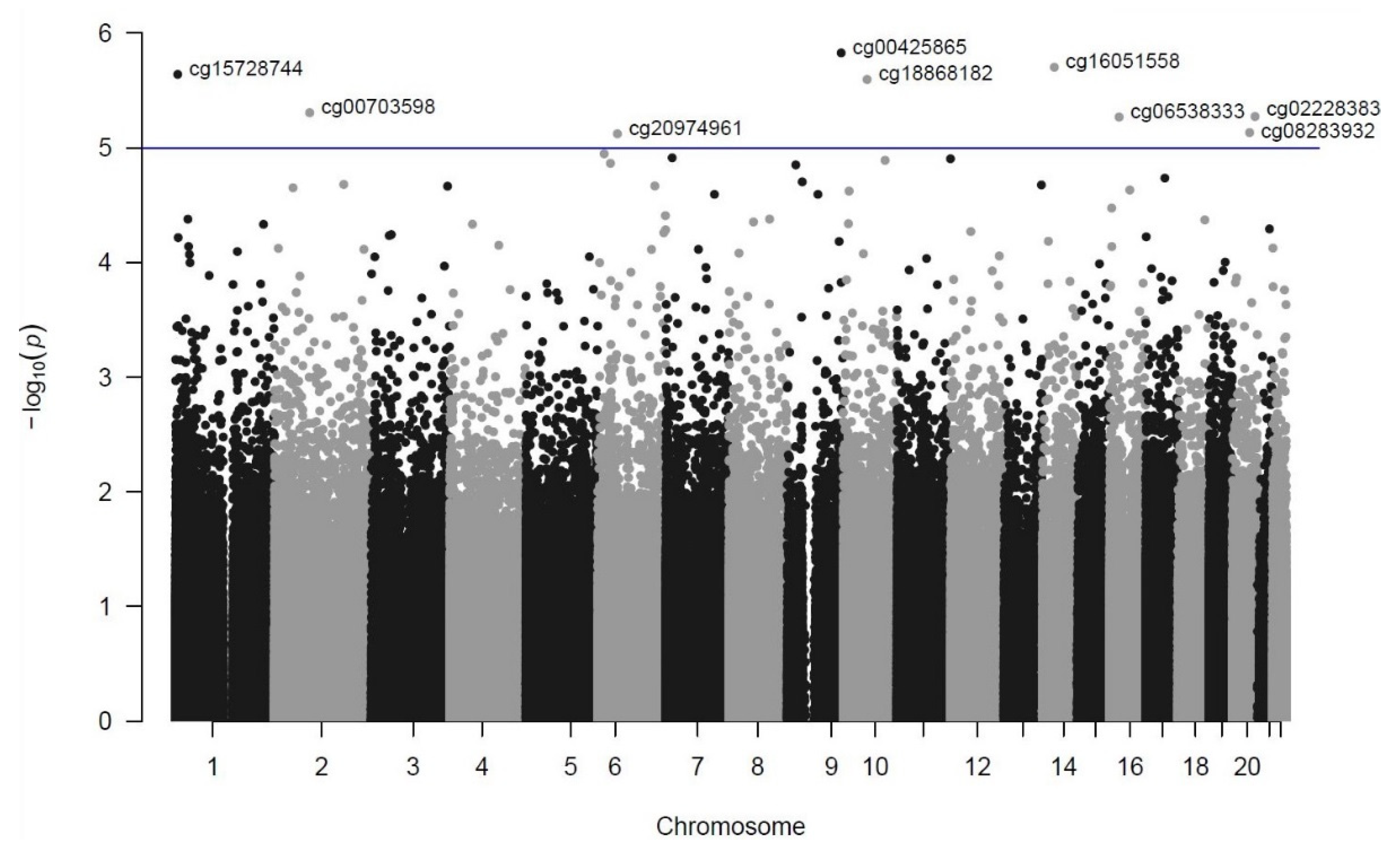
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Müller-Vahl, K.R.; Sambrani, T.; Jakubovski, E. Tic disorders revisited: Introduction of the term “tic spectrum disorders”. Eur. Child Adolesc. Psychiatry 2019, 28, 1129–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschtritt, M.E.; Lee, P.C.; Pauls, D.L.; Dion, Y.; Grados, M.A.; Illmann, C.; King, R.A.; Sandor, P.; McMahon, W.M.; Lyon, G.J.; et al. Lifetime prevalence, age of risk, and genetic relationships of comorbid psychiatric disorders in tourette syndrome. JAMA Psychiatry 2015, 72, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Burd, L.; Li, Q.; Kerbeshian, J.; Klug, M.G.; Freeman, R.D. Tourette syndrome and comorbid pervasive developmental disorders. J. Child Neurol. 2009, 24, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Mataix-Cols, D.; Isomura, K.; Pérez-Vigil, A.; Chang, Z.; Rück, C.; Johan Larsson, K.; Leckman, J.F.; Serlachius, E.; Larsson, H.; Lichtenstein, P. Familial risks of tourette syndrome and chronic tic disorders a population-based cohort study. JAMA Psychiatry 2015, 72, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.K.; Yu, D.; Keenan, C.L.; Gamazon, E.R.; Konkashbaev, A.I.; Derks, E.M.; Neale, B.M.; Yang, J.; Lee, S.H.; Evans, P.; et al. Partitioning the Heritability of Tourette Syndrome and Obsessive Compulsive Disorder Reveals Differences in Genetic Architecture. PLoS Genet. 2013, 9, e1003864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, B.; Melchior, L.; Jensen, L.R.; Groth, C.; Glenthøj, B.; Rizzo, R.; Debes, N.M.; Skov, L.; Brøndum-Nielsen, K.; Paschou, P.; et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur. J. Hum. Genet. 2014, 22, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, B.; Stefánsson, H.; Riff Jensen, L.; Melchior, L.; Mol Debes, N.; Groth, C.; Skov, L.; Werge, T.; Karagiannidis, I.; Tarnok, Z.; et al. Association of AADAC Deletion and Gilles de la Tourette Syndrome in a Large European Cohort. Biol. Psychiatry 2016, 79, 383–391. [Google Scholar] [CrossRef]
- Huang, A.Y.; Yu, D.; Davis, L.K.; Sul, J.H.; Tsetsos, F.; Ramensky, V.; Zelaya, I.; Ramos, E.M.; Osiecki, L.; Chen, J.A.; et al. Rare Copy Number Variants in NRXN1 and CNTN6 Increase Risk for Tourette Syndrome. Neuron 2017, 94, 1101–1111.e7. [Google Scholar] [CrossRef] [Green Version]
- Müller-Vahl, K.R.; Loeber, G.; Kotsiari, A.; Müller-Engling, L.; Frieling, H. Gilles de la Tourette syndrome is associated with hypermethylation of the dopamine D2 receptor gene. J. Psychiatr. Res. 2017, 86, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildonen, M.; Levy, A.M.; Dahl, C.; Bjerregaard, V.A.; Møller, L.B.; Guldberg, P.; Debes, N.M.; Tümer, Z. Elevated expression of SLC6A4 encoding the serotonin transporter (SERT) in gilles de la tourette syndrome. Genes 2021, 12, 86. [Google Scholar] [CrossRef]
- Tan, Q.; Christiansen, L.; Von Bornemann Hjelmborg, J.; Christensen, K. Twin methodology in epigenetic studies. J. Exp. Biol. 2015, 218, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Zilhão, N.R.; Padmanabhuni, S.S.; Pagliaroli, L.; Barta, C.; Smit, D.J.A.; Cath, D.; Nivard, M.G.; Baselmans, B.M.L.; Van Dongen, J.; Paschou, P.; et al. Epigenome-Wide Association Study of Tic Disorders. Twin Res. Hum. Genet. 2015, 18, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Salas, L.A.; Koestler, D.C.; Butler, R.A.; Hansen, H.M.; Wiencke, J.K.; Kelsey, K.T.; Christensen, B.C. FlowSorted.Blood.EPIC. DIM 2018, 575719, 289. [Google Scholar]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.S.; Schwartz, D.A.; Yang, I.V.; Kechris, K.J. Comb-p: Software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012, 28, 2986–2988. [Google Scholar] [CrossRef] [Green Version]
- Stessman, H.A.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.B.; Kigoshi-Tansho, Y.; Sher, R.B.; Ravenscroft, G.; Stauffer, J.E.; Kumar, R.; Yonashiro, R.; Müller, T.; Griffith, C.; Allen, W.; et al. NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat. Commun. 2020, 1, 1–12. [Google Scholar] [CrossRef]
- Stubbs, G.; Litt, M.; Lis, E.; Jackson, R.; Voth, W.; Lindberg, A.; Litt, R. Adenosine Deaminase Activity Decreased in Autism. J. Am. Acad. Child Psychiatry 1982, 21, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; De Luca, D.; Saccucci, P.; Fiumara, A.; Elia, M.; Porfirio, M.C.; Lucarelli, P.; Curatolo, P. Autism: Evidence of association with adenosine deaminase genetic polymorphism. Neurogenetics 2001, 3, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Duyzend, M.H.; Coe, B.P.; Baker, C.; Hoekzema, K.; Gerdts, J.; Turner, T.N.; Zody, M.C.; Beighley, J.S.; Murali, S.C.; et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet. Med. 2019, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.J.; Glessner, J.T.; Hakonarson, H. A Genome-Wide Association Study of Autism Incorporating Autism Diagnostic Interview-Revised, Autism Diagnostic Observation Schedule, and Social Responsiveness Scale. Child Dev. 2013, 84, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Nagai, M.; Mizuno, S.; Ogishima, S.; Tamiya, G.; Ueki, M.; Sakurai, R.; Makino, S.; Obara, T.; Ishikuro, M.; et al. Clustering by phenotype and genome-wide association study in autism. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.M.; Croen, L.A.; Torres, A.R.; Kharrazi, M.; Delorenze, G.N.; Windham, G.C.; Yoshida, C.K.; Zerbo, O.; Weiss, L.A. A Genome-Wide Survey of Transgenerational Genetic Effects in Autism. PLoS ONE 2013, 8, e76978. [Google Scholar] [CrossRef] [Green Version]
- Elia, J.; Gai, X.; Xie, H.M.; Perin, J.C.; Geiger, E.; Glessner, J.T.; D’Arcy, M.; Deberardinis, R.; Frackelton, E.; Kim, C.; et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 2010, 15, 637–646. [Google Scholar] [CrossRef]
- De Araújo Lima, L.; Feio-Dos-Santos, A.C.; Belangero, S.I.; Gadelha, A.; Bressan, R.A.; Salum, G.A.; Pan, P.M.; Moriyama, T.S.; Graeff-Martins, A.S.; Tamanaha, A.C.; et al. An integrative approach to investigate the respective roles of single-nucleotide variants and copy-number variants in Attention-Deficit/Hyperactivity Disorder. Sci. Rep. 2016, 6, 22851. [Google Scholar] [CrossRef]
- Wilson, M.M.; Henshall, D.C.; Byrne, S.M.; Brennan, G.P. Chd2-related cns pathologies. Int. J. Mol. Sci. 2021, 22, 588. [Google Scholar] [CrossRef]
- Egawa, J.; Watanabe, Y.; Wang, C.; Inoue, E.; Sugimoto, A.; Sugiyama, T.; Igeta, H.; Nunokawa, A.; Shibuya, M.; Kushima, I.; et al. Novel rare missense variations and risk of autism spectrum disorder: Whole-exome sequencing in two families with affected siblings and a two-stage follow-up study in a Japanese population. PLoS ONE 2015, 10, e0119413. [Google Scholar] [CrossRef]
- de Vries, P.J.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; Beaure d’Augères, G.; Ferreira, J.C.; et al. Tuberous Sclerosis Complex-Associated Neuropsychiatric Disorders (TAND): New Findings on Age, Sex, and Genotype in Relation to Intellectual Phenotype. Front. Neurol. 2020, 11, 603. [Google Scholar] [CrossRef]
- Tsetsos, F.; Padmanabhuni, S.S.; Alexander, J.; Karagiannidis, I.; Tsifintaris, M.; Topaloudi, A.; Mantzaris, D.; Georgitsi, M.; Drineas, P.; Paschou, P. Meta-analysis of tourette syndrome and attention deficit hyperactivity disorder provides support for a shared genetic basis. Front. Neurosci. 2016, 10, 340. [Google Scholar] [CrossRef]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 Complex Is Required for Proper Activation of mTOR Complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Saifetiarova, J.; Taylor, A.M.; Bhat, M.A. mTORC1 activation by loss of Tsc1 in myelinating glia causes downregulation of quaking and neurofascin 155 leading to paranodal domain disorganization. Front. Cell. Neurosci. 2018, 12, 201. [Google Scholar] [CrossRef]
- Kosillo, P.; Doig, N.M.; Ahmed, K.M.; Agopyan-Miu, A.H.C.W.; Wong, C.D.; Conyers, L.; Threlfell, S.; Magill, P.J.; Bateup, H.S. Tsc1-mTORC1 signaling controls striatal dopamine release and cognitive flexibility. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kitagishi, Y.; Kobayashi, M.; Kikuta, K.; Matsuda, S. Roles of PI3K/AKT/GSK3/mTOR pathway in cell signaling of mental illnesses. Depress. Res. Treat. 2012, 2012, 752563. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Mehan, S. Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 2021, 147, 105067. [Google Scholar] [CrossRef]
- Matsuda, S.; Ikeda, Y.; Murakami, M.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y. Roles of PI3K/AKT/GSK3 Pathway Involved in Psychiatric Illnesses. Diseases 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Y.; Yu, L.; Vickstrom, C.R.; Liu, Q.S. VTA mTOR Signaling Regulates Dopamine Dynamics, Cocaine-Induced Synaptic Alterations, and Reward. Neuropsychopharmacology 2018, 43, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Buse, J.; Schoenefeld, K.; Münchau, A.; Roessner, V. Neuromodulation in Tourette syndrome: Dopamine and beyond. Neurosci. Biobehav. Rev. 2013, 37, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Maia, T.V.; Conceição, V.A. Dopaminergic Disturbances in Tourette Syndrome: An Integrative Account. Biol. Psychiatry 2018, 84, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, H.; Balasubramanian, V.; Iyer, M.; Venugopal, A.; Subramaniam, M.D.; Cho, S.G.; Vellingiri, B. mTOR signalling pathway—A root cause for idiopathic autism? BMB Rep. 2019, 52, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururajan, A.; Van Den Buuse, M. Is the mTOR-signalling cascade disrupted in Schizophrenia? J. Neurochem. 2014, 129, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Chadha, R.; Meador-Woodruff, J.H. Downregulated AKT-mTOR signaling pathway proteins in dorsolateral prefrontal cortex in Schizophrenia. Neuropsychopharmacology 2020, 45, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Abelaira, H.M.; Réus, G.Z.; Neotti, M.V.; Quevedo, J. The role of mTOR in depression and antidepressant responses. Life Sci. 2014, 101, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.F.; Yu, J.T.; Song, J.H.; Chi, S.; Tan, L. Role of the mTOR signaling pathway in epilepsy. J. Neurol. Sci. 2013, 332, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.C.; Huang, H.L.; Weng, W.C.; Jong, Y.J.; Yin, Y.J.; Chen, H.A.; Lee, W.T.; Ho, S.Y. Increased risk of epilepsy in children with Tourette syndrome: A population-based case-control study. Res. Dev. Disabil. 2016, 51–52, 181–187. [Google Scholar] [CrossRef]
- Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; Malik, R.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Drakos, E.; Grammatikakis, I.; Schlette, E.J.; Li, J.; Leventaki, V.; Staikou-Drakopoulou, E.; Patsouris, E.; Panayiotidis, P.; Medeiros, L.J.; et al. MTOR signaling is activated by FLT3 kinase and promotes survival of FLT3-mutated acute myeloid leukemia cells. Mol. Cancer 2010, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Tsetsos, F.; Yu, D.; Sul, J.H.; Huang, A.Y.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; Greenberg, E.; Muller-Vahl, K.R.; et al. Synaptic processes and immune-related pathways implicated in Tourette syndrome. Transl. Psychiatry 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Yu, D.; Sul, J.H.; Tsetsos, F.; Nawaz, M.S.; Huang, A.Y.; Zelaya, I.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; et al. Interrogating the genetic determinants of Tourette’s syndrome and other tiC disorders through genome-wide association studies. Am. J. Psychiatry 2019, 176, 217–227. [Google Scholar] [CrossRef]
- Eriguchi, Y.; Kuwabara, H.; Inai, A.; Kawakubo, Y.; Nishimura, F.; Kakiuchi, C.; Tochigi, M.; Ohashi, J.; Aoki, N.; Kato, K.; et al. Identification of candidate genes involved in the etiology of sporadic Tourette syndrome by exome sequencing. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 712–723. [Google Scholar] [CrossRef]
- Qi, Q.; Menzaghi, C.; Smith, S.; Liang, L.; de Rekeneire, N.; Garcia, M.E.; Lohman, K.K.; Miljkovic, I.; Strotmeyer, E.S.; Cummings, S.R.; et al. Genome-wide association analysis identifies TYW3/CRYZ and NDST4 loci associated with circulating resistin levels. Hum. Mol. Genet. 2012, 21, 4774–4780. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Chemaly, E.R.; Hajjar, R.J.; Lebeche, D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J. Biol. Chem. 2011, 286, 18465–18473. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Rank | DMP (CpG Site) | Chromosome Position (GRCh37/hg19) | p-Value | FDR | Coefficient | Closest Genes (Distance from TSS in bp) a |
|---|---|---|---|---|---|---|
| 1 | cg00425865 | chr9:135820111 | 1.49 × 10−6 | 0.48 | −0.6889 | TSC1 (−104)/GFI1B (−826) |
| 2 | cg16051558 | chr14:50235537 | 1.98 × 10−6 | 0.48 | 0.3805 | KLHDC2 (+1212)/NEMF (+84,383) |
| 3 | cg15728744 | chr1:5917533 | 2.29 × 10−6 | 0.48 | 0.5383 | NPHP4 (+134,997) |
| 4 | cg18868182 | chr10:60086874 | 2.54 × 10−6 | 0.48 | 0.1460 | UBE2D1 (−7860)/CISD1 (+58,057) |
| 5 | cg00703598 | chr2:88480233 | 4.94 × 10−6 | 0.58 | −0.2805 | THNSL2 (+9255)/TEX37 (−343,935) |
| 6 | cg02228383 | chr20:56934684 | 5.33 × 10−6 | 0.58 | 0.5032 | VAPB (−29,493)/RAB22A (+49,933) |
| 7 | cg06538333 | chr16:23445078 | 5.39 × 10−6 | 0.58 | −0.6260 | COG7 (+19,422)/SCNN1B (+131,488) |
| 8 | cg08283932 | chr20:43280707 | 7.36 × 10−6 | 0.63 | −0.6783 | ADA (−325) |
| 9 | cg20974961 | chr6:49500569 | 7.54 × 10−6 | 0.63 | −0.3322 | GLYATL3 (+32,899)/RHAG (+103,982) |
| Ontology | Term | Binom Raw p-Value | Binom FDR q-Value | Binom Fold Enrichment | Binom Observed Region Hits |
|---|---|---|---|---|---|
| GO Cellular Component | Insulin receptor substrate binding | 3.88 × 10−4 | 4.33 × 10−2 | 2.25 | 23 |
| GO Biological Process | Positive regulation of hormone metabolic process | 9.20 × 10−6 | 1.12 × 10−3 | 2.70 | 26 |
| Positive regulation of hormone biosynthetic process | 1.56 × 10−5 | 1.79 × 10−3 | 3.08 | 20 | |
| Regulation of protein ubiquitination involved in ubiquitin-dependent protein catabolic process | 5.10 × 10−4 | 2.65 × 10−2 | 2.36 | 20 | |
| MSigDB Pathway | Phosphoinositides and their downstream targets | 7.62 × 10−4 | 3.14 × 10−2 | 2.07 | 25 |
| PTEN is a tumor suppressor that dephosphorylates the lipid messenger phosphatidylinositol triphosphate | 1.13 × 10−3 | 3.73 × 10−2 | 2.39 | 17 |
| Rank | Chromosome Position (GRCh37/hg19) | Length (bp) | No. of Sites | SLK adj. p-Value | Šidák adj. p-Value | Closest Genes (Distance to TSS) a |
|---|---|---|---|---|---|---|
| 1 | chr1:75198768-75199178 | 410 | 8 | 7.60 × 10−6 | 0.001 | CRYZ (+119)/TYW3 (+133) |
| 2 | chr7:3227262-3227333 | 71 | 3 | 9.01 × 10−6 | 0.091 | SDK1 (−113,782)/CARD11 (−143,719) |
| 3 | chr3:138067848-138068014 | 166 | 6 | 3.07 × 10−5 | 0.131 | MRAS (+1392)/ESYT3 (−85,524) |
| 4 | chr15:93353059-93353199 | 140 | 3 | 7.35 × 10−5 | 0.328 | CHD2 (−89,929)/FAM174B (−153,941) |
| 5 | chr19:17830341-17830453 | 112 | 3 | 7.61 × 10−5 | 0.402 | MAP1S (+236) |
| 6 | chr13:41635362-41635513 | 151 | 4 | 1.05 × 10−4 | 0.409 | WBP4 (+28) |
| 7 | chr8:1765217-1765388 | 171 | 7 | 1.54 × 10−4 | 0.494 | ARHGEF10 (−6839)/CLN8 (+53,375) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hildonen, M.; Levy, A.M.; Hansen, C.S.; Bybjerg-Grauholm, J.; Skytthe, A.; Debes, N.M.; Tan, Q.; Tümer, Z. EWAS of Monozygotic Twins Implicate a Role of mTOR Pathway in Pathogenesis of Tic Spectrum Disorder. Genes 2021, 12, 1510. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101510
Hildonen M, Levy AM, Hansen CS, Bybjerg-Grauholm J, Skytthe A, Debes NM, Tan Q, Tümer Z. EWAS of Monozygotic Twins Implicate a Role of mTOR Pathway in Pathogenesis of Tic Spectrum Disorder. Genes. 2021; 12(10):1510. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101510
Chicago/Turabian StyleHildonen, Mathis, Amanda M. Levy, Christine Søholm Hansen, Jonas Bybjerg-Grauholm, Axel Skytthe, Nanette M. Debes, Qihua Tan, and Zeynep Tümer. 2021. "EWAS of Monozygotic Twins Implicate a Role of mTOR Pathway in Pathogenesis of Tic Spectrum Disorder" Genes 12, no. 10: 1510. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101510