
Comparative Transcriptome Analysis in Monocyte-Derived Macrophages of Asymptomatic GBA Mutation Carriers and Patients with GBA-Associated Parkinson’s Disease

Abstract
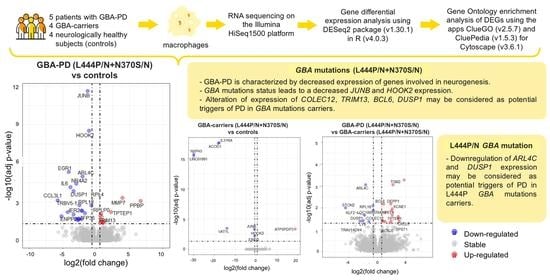
:
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Differentiation of Human Monocytes to Macrophages
2.3. RNA Isolation and RNA Sequencing (RNA-Seq)
2.4. Quality Control
2.5. Reads Mapping to Reference Genome
2.6. Quantification of Gene Expression Level
2.7. Analysis of Gene Differential Expression
2.8. GO Enrichment Analysis of Differentially Expressed Genes
3. Results
3.1. RNA-Seq Experiments
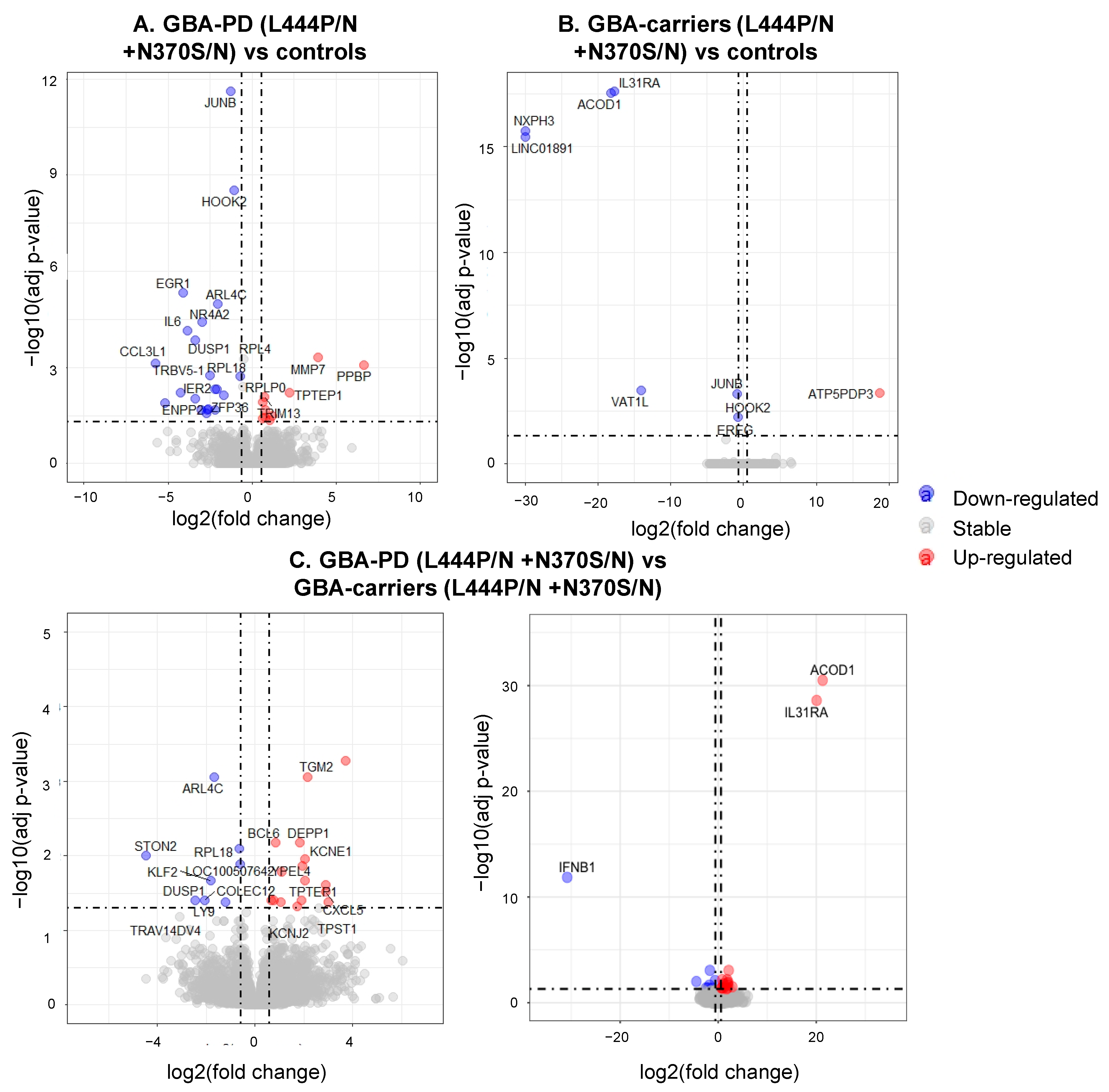
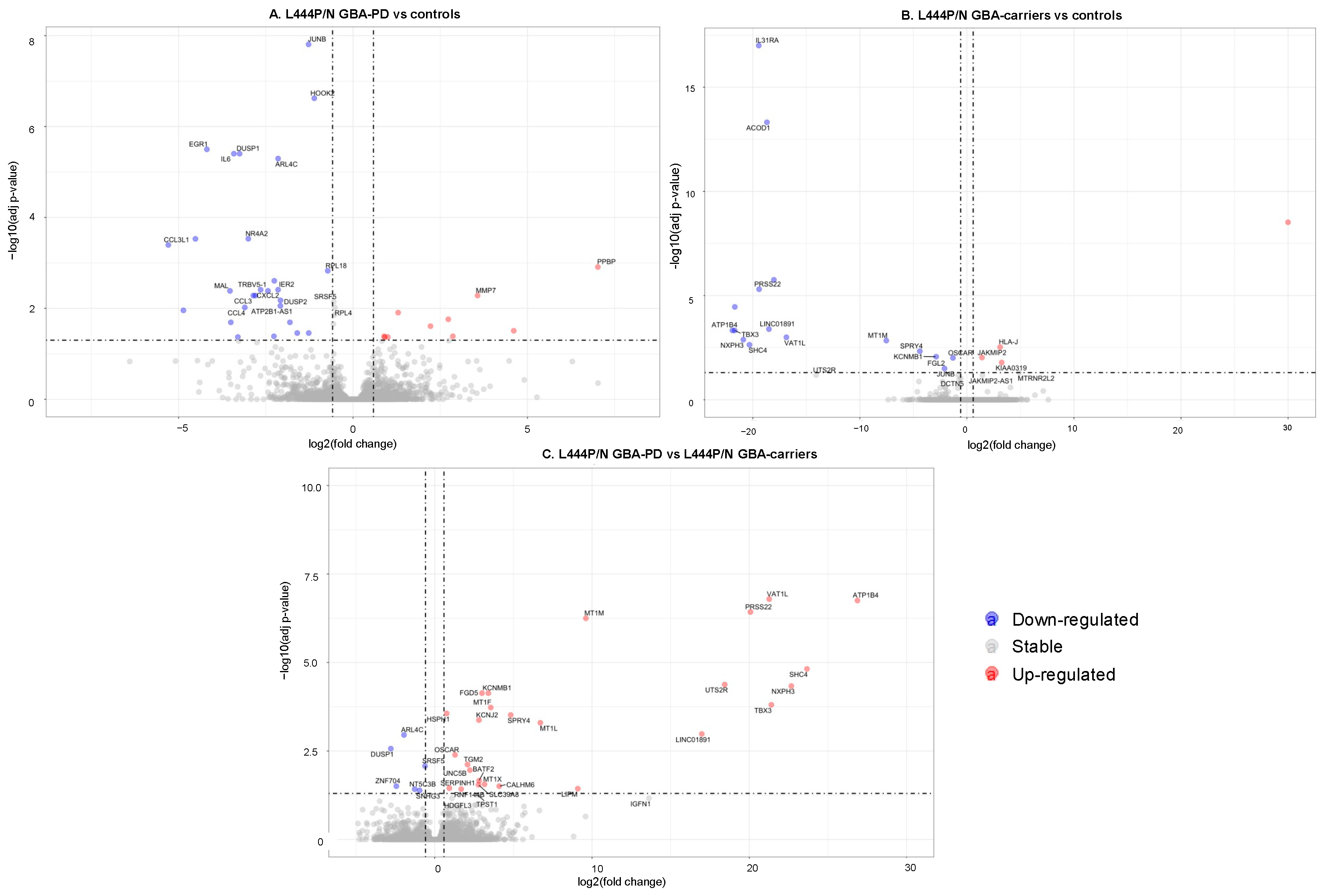
3.2. Changes in the Transcriptome Attributed to the Presence of GBA L444P/N Mutation
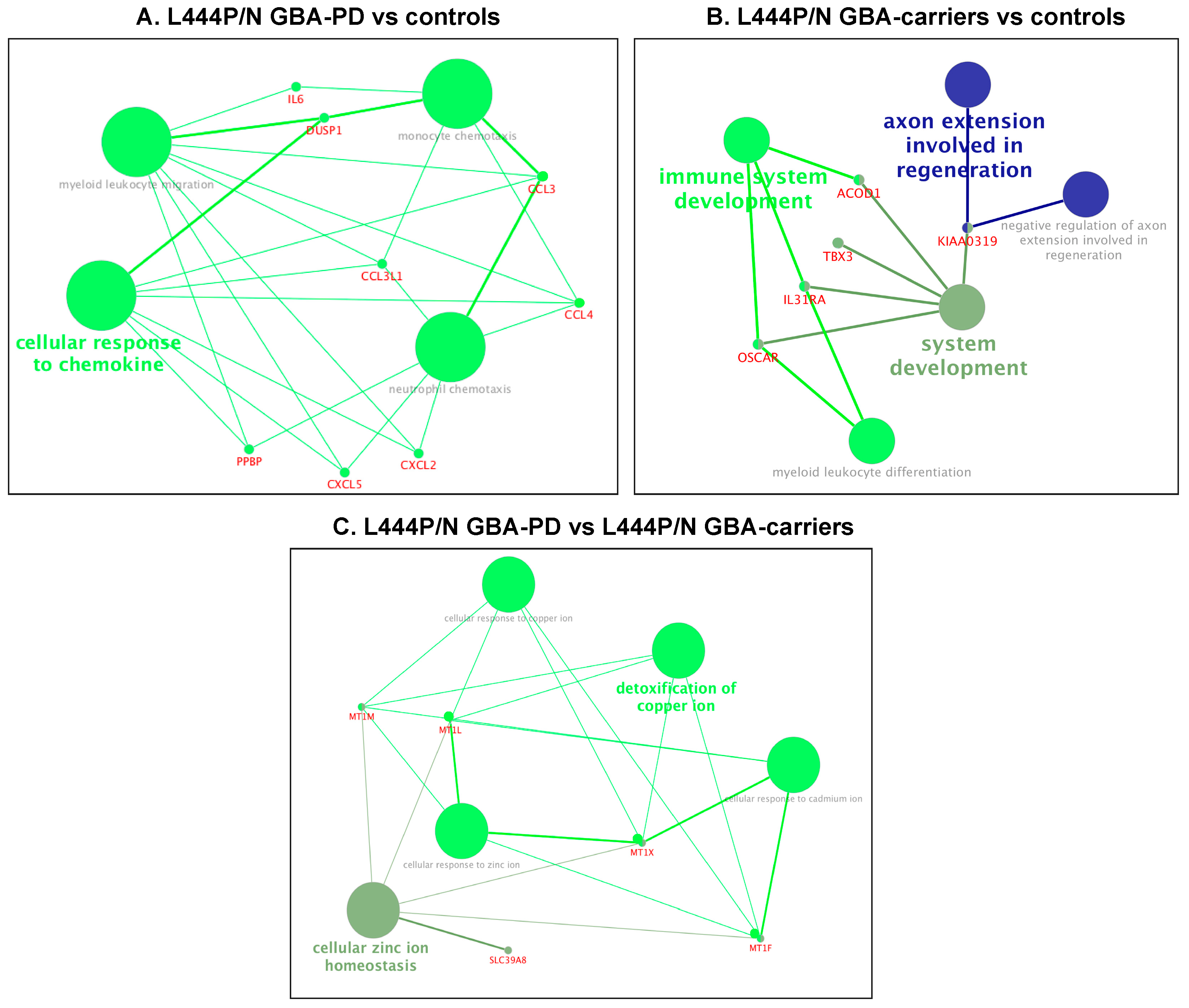
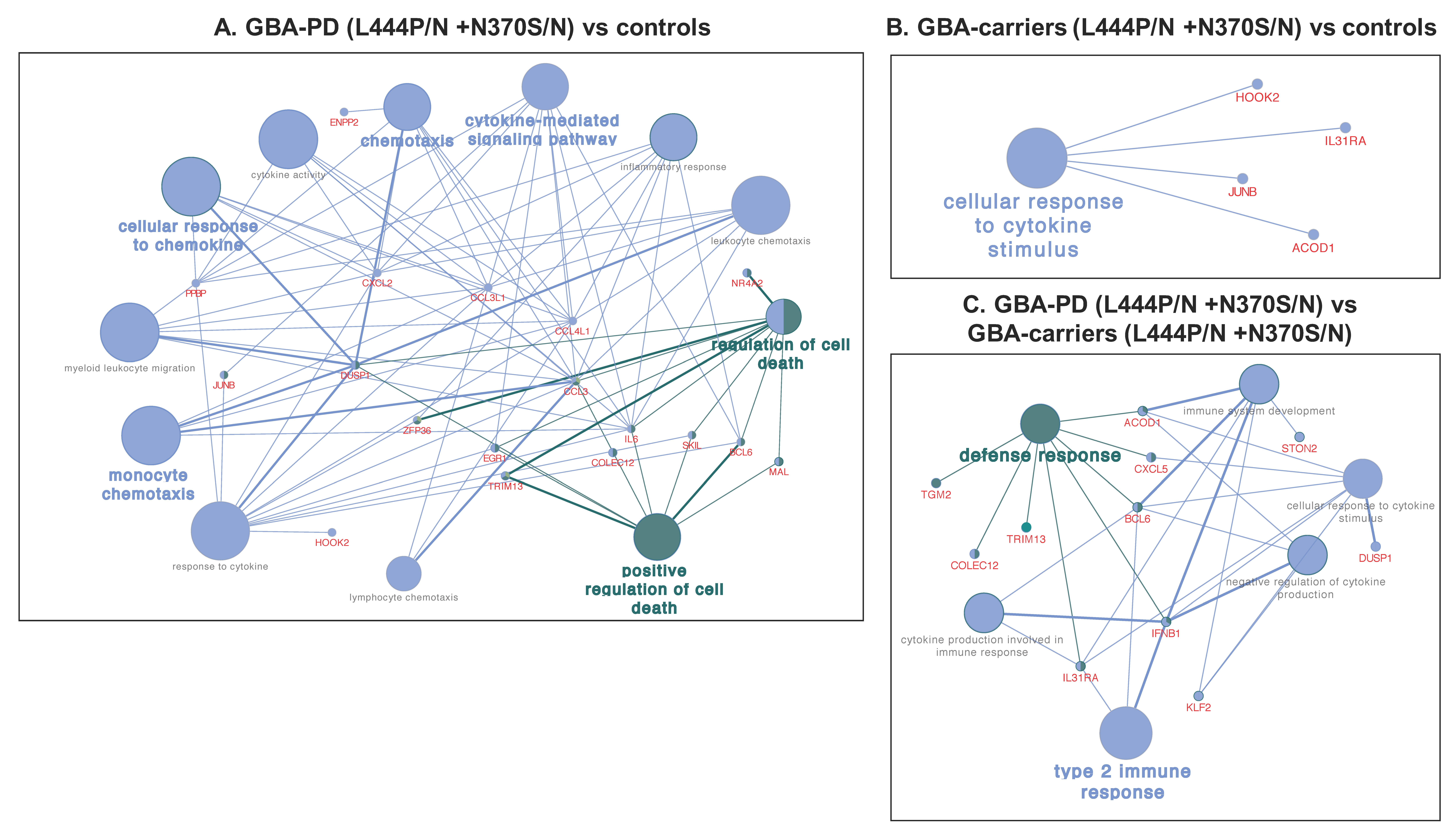
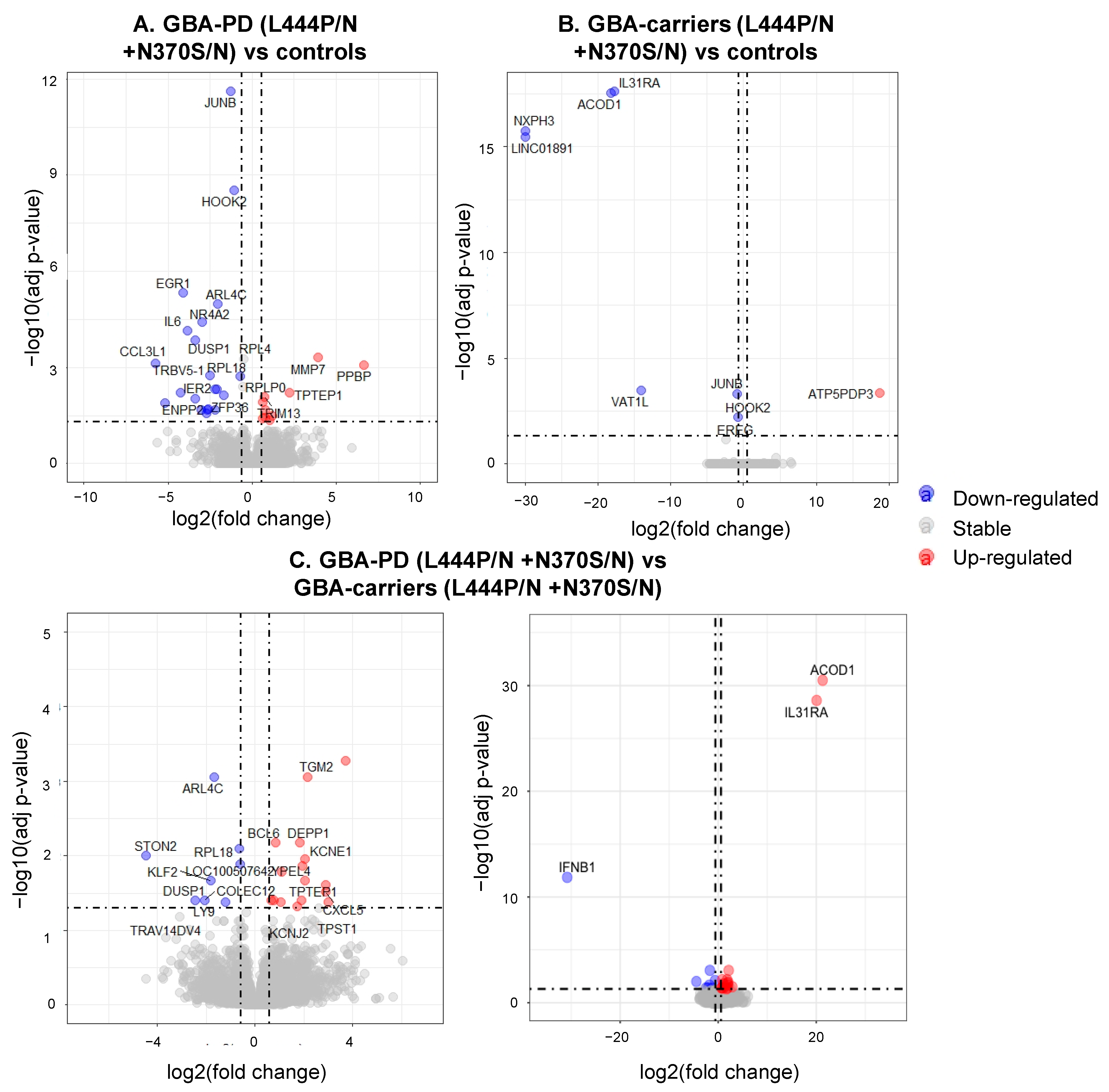
3.3. Differentially Expressed Genes and Enriched Pathways in GBA-PD Patients (L444P/N +N370S/N) and GBA Carriers (L444P/N +N370S/N) Compared to Controls
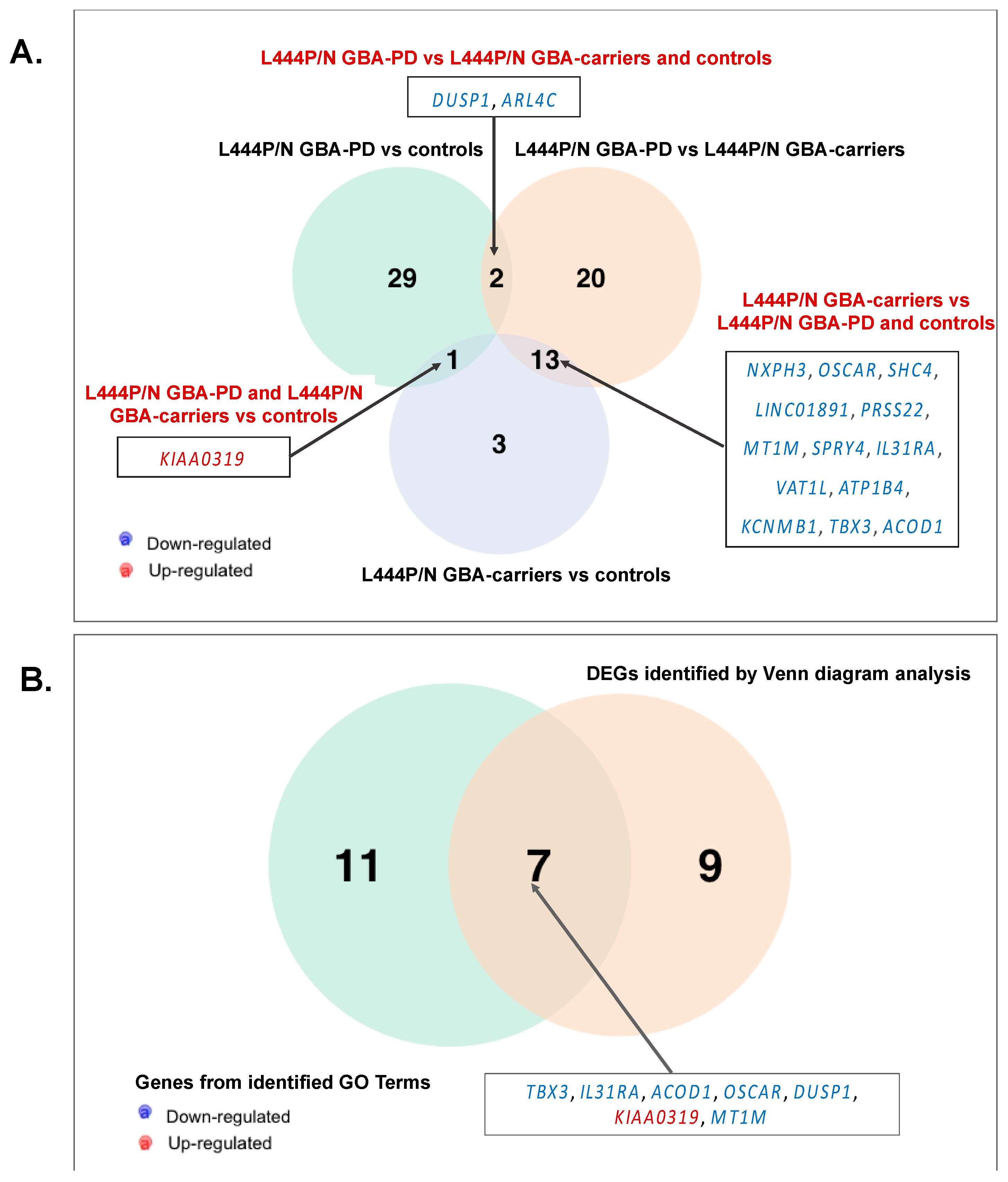
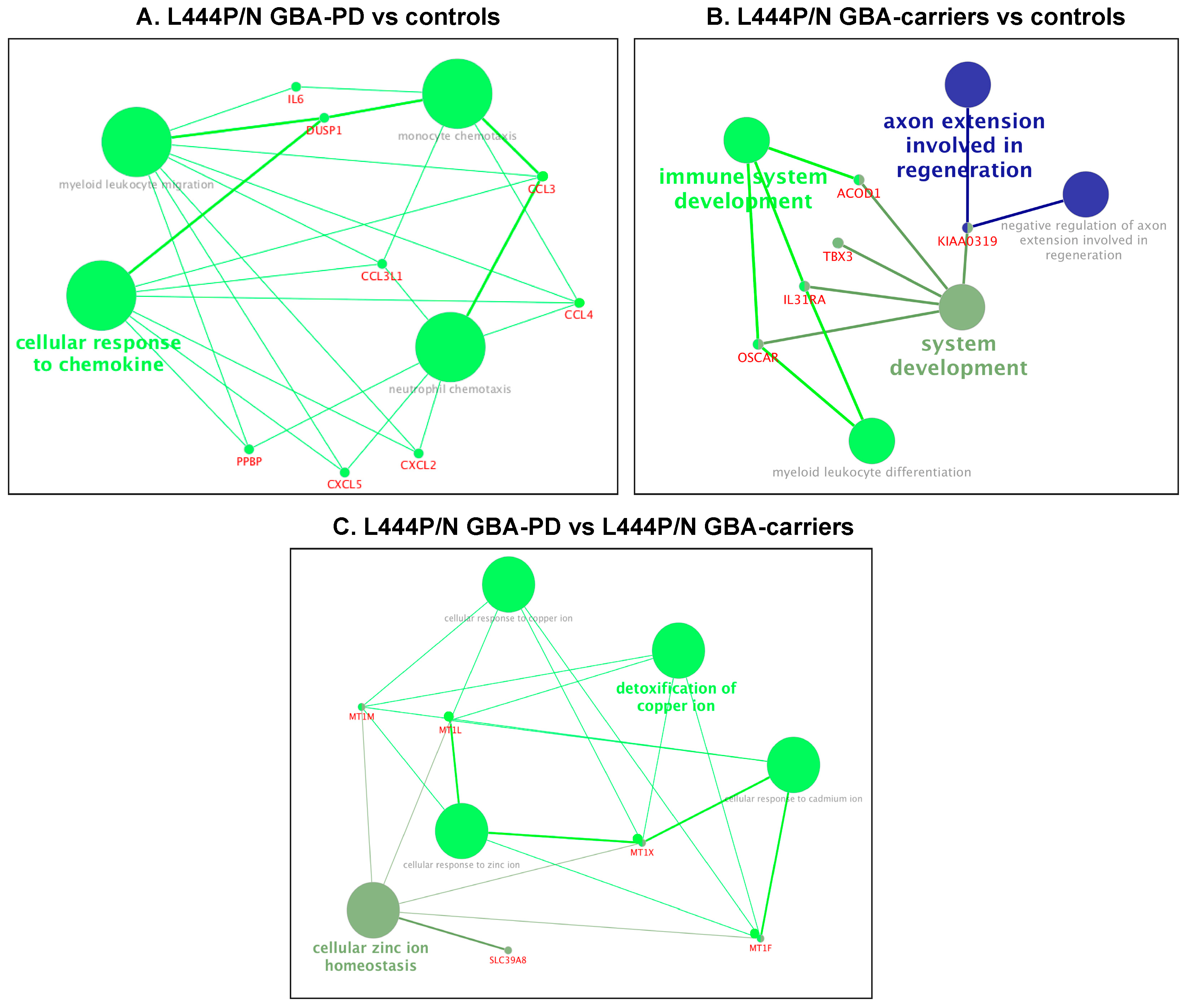
3.4. Differential Expression of Genes and Pathways in GBA-PD Patients (L444P/N +N370S/N) and GBA Carriers (L444P/N +N370S/N)
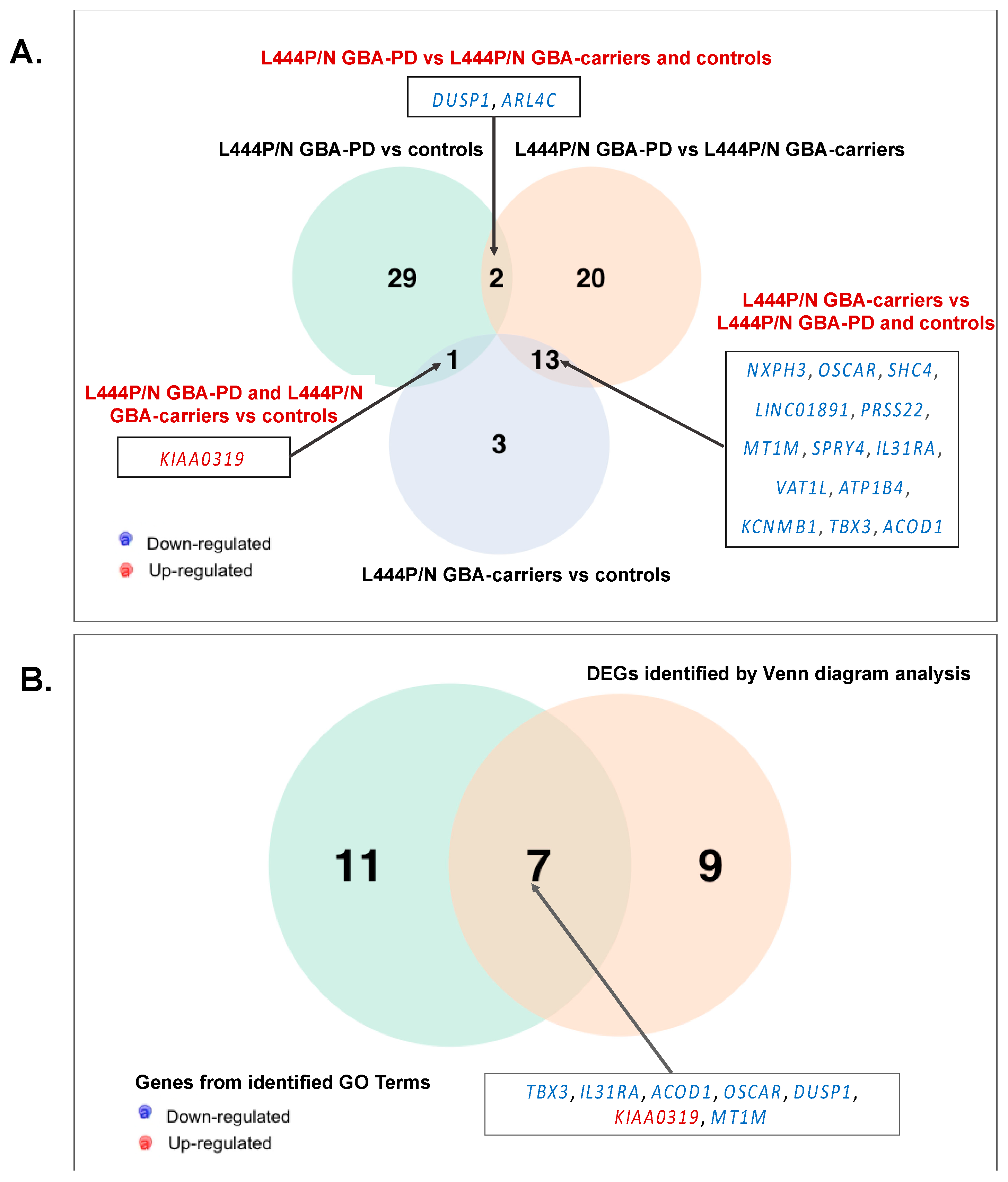
3.5. Searching the Overlapping DEGs between our and Publicly Available Dataset
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Parkinson's Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emelyanov, A.K.; Usenko, T.S.; Tesson, C.; Senkevich, K.A.; Nikolaev, M.A.; Miliukhina, I.V.; Kopytova, A.E.; Timofeeva, A.A.; Yakimovsky, A.F.; Lesage, S.; et al. Mutation analysis of Parkinson’s disease genes in a Russian data set. Neurobiol. Aging 2018, 71, 267-e7. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.-C.; Honoré, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 202–210. [Google Scholar] [CrossRef]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Blumenreich, S.; Barav, O.B.; Jenkins, B.J.; Futerman, A.H. Lysosomal Storage Disorders Shed Light on Lysosomal Dysfunction in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4966. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alcalay, R.N.; et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef]
- Pchelina, S.; Emelyanov, A.; Baydakova, G.; Andoskin, P.; Senkevich, K.; Nikolaev, M.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Fedotova, E.; et al. Oligomeric α-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson’s disease. Neurosci. Lett. 2017, 636, 70–76. [Google Scholar] [CrossRef]
- Pchelina, S.; Baydakova, G.; Nikolaev, M.; Senkevich, K.; Emelyanov, A.; Kopytova, A.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Berkovich, O.; et al. Blood lysosphingolipids accumulation in patients with parkinson’s disease with glucocerebrosidase 1 mutations. Mov. Disord. 2018, 33, 1325–1330. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Kirby, J.; Ferraiuolo, L.; Heath, P.R.; Rattray, M.; Shaw, P.J. Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 2012, 8, 518–530. [Google Scholar] [CrossRef]
- Mutez, E.; Nkiliza, A.; Belarbi, K.; de Broucker, A.; Vanbesien-Mailliot, C.; Bleuse, S.; Duflot, A.; Comptdaer, T.; Semaille, P.; Blervaque, R.; et al. Involvement of the immune system, endocytosis and EIF2 signaling in both genetically determined and sporadic forms of Parkinson’s disease. Neurobiol. Dis. 2014, 63, 165–170. [Google Scholar] [CrossRef]
- Infante, J.; Prieto, C.; Sierra, M.; Sánchez-Juan, P.; González-Aramburu, I.; Sánchez-Quintana, C.; Berciano, J.; Combarros, O.; Sainz, J. Identification of candidate genes for Parkinson’s disease through blood transcriptome analysis in LRRK2-G2019S carriers, idiopathic cases, and controls. Neurobiol. Aging 2015, 36, 1105–1109. [Google Scholar] [CrossRef]
- Infante, J.; Prieto, C.; Sierra, M.; Sánchez-Juan, P.; González-Aramburu, I.; Sánchez-Quintana, C.; Berciano, J.; Combarros, O.; Sainz, J. Comparative blood transcriptome analysis in idiopathic and LRRK2 G2019S-associated Parkinson’s disease. Neurobiol. Aging 2016, 38, 214.e1–214.e5. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Stubblefield, B.K.; Maniwang, E.; Lopez, G.; Moaven, N.; Goldin, E.; Marugan, J.; Patnaik, S.; Dutra, A.; Southall, N.; et al. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci. Transl. Med. 2014, 6, 240ra73. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Grabowski, G.A. Immunological cells and functions in Gaucher disease. Crit. Rev. Oncog. 2013, 18, 197–220. [Google Scholar] [CrossRef]
- Kopytova, A.E.; Rychkov, G.N.; Nikolaev, M.A.; Baydakova, G.V.; Cheblokov, A.A.; Senkevich, K.A.; Bogdanova, D.A.; Bolshakova, O.I.; Miliukhina, I.V.; Bezrukikh, V.A.; et al. Ambroxol increases glucocerebrosidase (GCase) activity and restores GCase translocation in primary patient-derived macrophages in Gaucher disease and Parkinsonism. Parkinsonism Relat. Disord. 2021, 84, 112–121. [Google Scholar] [CrossRef]
- Welsh, N.J.; Gewinner, C.A.; Mistry, K.; Koglin, M.; Cooke, J.; Butler, M.; Powney, B.; Roberts, M.; Staddon, J.M.; Schapira, A.H. V Functional assessment of glucocerebrosidase modulator efficacy in primary patient-derived macrophages is essential for drug development and patient stratification. Haematologica 2020, 105, e206–e209. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Nikolaev, M.A.; Kopytova, A.E.; Baidakova, G.V.; Emel’yanov, A.K.; Salogub, G.N.; Senkevich, K.A.; Usenko, T.S.; Gorchakova, M.V.; Koval’chuk, Y.P.; Berkovich, O.A.; et al. Human Peripheral Blood Macrophages as a Model for Studying Glucocerebrosidase Dysfunction. Cell Tissue Biol. 2019, 13, 100–106. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 9 September 2021).
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Borrageiro, G.; Haylett, W.; Seedat, S.; Kuivaniemi, H.; Bardien, S. A review of genome-wide transcriptomics studies in Parkinson’s disease. Eur. J. Neurosci. 2018, 47, 1–16. [Google Scholar] [CrossRef]
- Botta-Orfila, T.; Sànchez-Pla, A.; Fernández, M.; Carmona, F.; Ezquerra, M.; Tolosa, E. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson’s disease. Brain Res. 2012, 1466, 152–157. [Google Scholar] [CrossRef]
- Pallos, J.; Jeng, S.; McWeeney, S.; Martin, I. Dopamine neuron-specific LRRK2 G2019S effects on gene expression revealed by translatome profiling. Neurobiol. Dis. 2021, 155, 105390. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Lang, C.; Campbell, K.R.; Ryan, B.J.; Carling, P.; Attar, M.; Vowles, J.; Perestenko, O.V.; Bowden, R.; Baig, F.; Kasten, M.; et al. Single-Cell Sequencing of iPSC-Dopamine Neurons Reconstructs Disease Progression and Identifies HDAC4 as a Regulator of Parkinson Cell Phenotypes. Cell Stem Cell 2019, 24, 93–106.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.-Y.; Kho, D.; Reiners, J.J.J.; Wu, G.S. Role for DUSP1 (dual-specificity protein phosphatase 1) in the regulation of autophagy. Autophagy 2016, 12, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Bassil, K.; Al Jowf, G.I.; Steinbusch, H.W.M.; Rothermel, M.; de Nijs, L.; Rutten, B.P.F. Dual-specificity phosphatases in mental and neurological disorders. Prog. Neurobiol. 2021, 198, 101906. [Google Scholar] [CrossRef] [PubMed]
- Bhore, N.; Wang, B.-J.; Chen, Y.-W.; Liao, Y.-F. Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases. Int. J. Mol. Sci. 2017, 18, 1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.; Walczak, R.; Dhamko, H.; Bradley, M.N.; Marathe, C.; Boyadjian, R.; Salazar, J.V.; Tontonoz, P. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: Identification of ARL7 as a direct target. J. Lipid Res. 2011, 52, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, R.; Landreth, G.E. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol. Metab. 2016, 27, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, P.; Kaufmann, W.E.; Galaburda, A.M. Developmental dyslexia in women: Neuropathological findings in three patients. Ann. Neurol. 1990, 28, 727–738. [Google Scholar] [CrossRef]
- Galaburda, A.M.; LoTurco, J.; Ramus, F.; Fitch, R.H.; Rosen, G.D. From genes to behavior in developmental dyslexia. Nat. Neurosci. 2006, 9, 1213–1217. [Google Scholar] [CrossRef]
- Tomar, D.; Singh, R.; Singh, A.K.; Pandya, C.D.; Singh, R. TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim. Biophys. Acta 2012, 1823, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef] [PubMed]
- Narayan, K.; Waggoner, L.; Pham, S.T.; Hendricks, G.L.; Waggoner, S.N.; Conlon, J.; Wang, J.P.; Fitzgerald, K.A.; Kang, J. TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J. Virol. 2014, 88, 10748–10757. [Google Scholar] [CrossRef] [Green Version]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, L.; Skjodt, K.; Nielsen, O.; Floridon, C.; Holmskov, U.; Hansen, S. Expression and tissue localization of collectin placenta 1 (CL-P1, SRCL) in human tissues. Mol. Immunol. 2008, 45, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- Kutyavin, V.I.; Chawla, A. BCL6 regulates brown adipocyte dormancy to maintain thermogenic reserve and fitness. Proc. Natl. Acad. Sci. USA 2019, 116, 17071–17080. [Google Scholar] [CrossRef] [Green Version]
- Bogie, J.F.J.; Mailleux, J.; Wouters, E.; Jorissen, W.; Grajchen, E.; Vanmol, J.; Wouters, K.; Hellings, N.; van Horssen, J.; Vanmierlo, T.; et al. Scavenger receptor collectin placenta 1 is a novel receptor involved in the uptake of myelin by phagocytes. Sci. Rep. 2017, 7, 44794. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, L.; Wiegmann, C.; Arsalan-Werner, A.; Hilbrich, I.; Jäger, C.; Flach, K.; Suttkus, A.; Lachmann, I.; Arendt, T.; Holzer, M. Hook proteins: Association with Alzheimer pathology and regulatory role of hook3 in amyloid beta generation. PLoS ONE 2015, 10, e0119423. [Google Scholar] [CrossRef] [Green Version]
- Szebenyi, G.; Wigley, W.C.; Hall, B.; Didier, A.; Yu, M.; Thomas, P.; Krämer, H. Hook2 contributes to aggresome formation. BMC Cell Biol. 2007, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walenta, J.H.; Didier, A.J.; Liu, X.; Krämer, H. The Golgi-associated hook3 protein is a member of a novel family of microtubule-binding proteins. J. Cell Biol. 2001, 152, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakaria, M.; Haque, M.E.; Cho, D.-Y.; Azam, S.; Kim, I.-S.; Choi, D.-K. Molecular Insights into NR4A2(Nurr1): An Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death. Mol. Neurobiol. 2019, 56, 5799–5814. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Wang, Y.; Paudel, H.K. Early Growth Response 1 (Egr-1) Is a Transcriptional Activator of β-Secretase 1 (BACE-1) in the Brain. J. Biol. Chem. 2016, 291, 22276–22287. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, Y.; Ikeda, T.; Saito-Takatsuji, H.; Yonekura, H. Emerging Role of AP-1 Transcription Factor JunB in Angiogenesis and Vascular Development. Int. J. Mol. Sci. 2021, 22, 2804. [Google Scholar] [CrossRef] [PubMed]
- Zukin, R.; Jover-Mengual, T.; Yokota, H.; Calderone, A.; Simionescu, M.; Lau, C.G. Molecular and Cellular Mechanisms of Ischemia-Induced Neuronal Death. Stroke Pathophysiol. Diagn. Manag. 2004. [Google Scholar] [CrossRef]
- Lee, J.K.H.; Pearson, J.D.; Maser, B.E.; Ingham, R.J. Cleavage of the JunB transcription factor by caspases generates a carboxyl-terminal fragment that inhibits activator protein-1 transcriptional activity. J. Biol. Chem. 2013, 288, 21482–21495. [Google Scholar] [CrossRef] [Green Version]
- Trizzino, M.; Zucco, A.; Deliard, S.; Wang, F.; Barbieri, E.; Veglia, F.; Gabrilovich, D.; Gardini, A. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci. Adv. 2021, 7, eaaz8836. [Google Scholar] [CrossRef]
- Winter, C.; Weiss, C.; Martin-Villalba, A.; Zimmermann, M.; Schenkel, J. JunB and Bcl-2 overexpression results in protection against cell death of nigral neurons following axotomy. Brain Res. Mol. Brain Res. 2002, 104, 194–202. [Google Scholar] [CrossRef]
- Fontana, M.F.; Baccarella, A.; Pancholi, N.; Pufall, M.A.; Herbert, D.R.; Kim, C.C. JUNB is a key transcriptional modulator of macrophage activation. J. Immunol. 2015, 194, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Huang, Q.; Du, X.; Xu, S.; Li, M.; Ma, S. Early activation of Egr-1 promotes neuroinflammation and dopaminergic neurodegeneration in an experimental model of Parkinson’s disease. Exp. Neurol. 2018, 302, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Chen, S.; Le, W.D. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog. Neurobiol. 2005, 77, 128–138. [Google Scholar] [CrossRef]
- Le, W.-D.; Xu, P.; Jankovic, J.; Jiang, H.; Appel, S.H.; Smith, R.G.; Vassilatis, D.K. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 2003, 33, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, H.; Li, T.; Cui, J.; Fu, Y.; Ren, J.; Sun, X.; Jiang, P.; Yu, S.; Li, C. NR4A2 genetic variation and Parkinson’s disease: Evidence from a systematic review and meta-analysis. Neurosci. Lett. 2017, 650, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, P.M.A.; Healy, D.G.; Muqit, M.M.K.; Yang, Y.X.; Van Der Brug, M.; Holton, J.L.; Revesz, T.; Quinn, N.P.; Bhatia, K.; Diss, J.K.J.; et al. Characterisation of a novel NR4A2 mutation in Parkinson’s disease brain. Neurosci. Lett. 2009, 457, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Sánchez, E.; Yescas, P.; Rodríguez-Violante, M.; Martínez-Rodríguez, N.; Díaz-López, J.N.; Ochoa, A.; Valdes-Rojas, S.S.; Magos-Rodríguez, D.; Rojas-Castañeda, J.C.; Cervantes-Arriaga, A.; et al. Association of polymorphisms and reduced expression levels of the NR4A2 gene with Parkinson’s disease in a Mexican population. J. Neurol. Sci. 2017, 379, 58–63. [Google Scholar] [CrossRef]
- Mahajan, S.; Saini, A.; Chandra, V.; Nanduri, R.; Kalra, R.; Bhagyaraj, E.; Khatri, N.; Gupta, P. Nuclear Receptor Nr4a2 Promotes Alternative Polarization of Macrophages and Confers Protection in Sepsis. J. Biol. Chem. 2015, 290, 18304–18314. [Google Scholar] [CrossRef] [Green Version]
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 831. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, K.; Schulte, C.; Schneiderhan-Marra, N.; Apel, A.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.-C.; Cormier, F.; et al. Inflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur. J. Neurol. 2017, 24, 427-e6. [Google Scholar] [CrossRef]
- Miliukhina, I.V.; Usenko, T.S.; Senkevich, K.A.; Nikolaev, M.A.; Timofeeva, A.A.; Agapova, E.A.; Semenov, A.V.; Lubimova, N.E.; Totolyan, A.A.; Pchelina, S.N. Plasma Cytokines Profile in Patients with Parkinson’s Disease Associated with Mutations in GBA Gene. Bull. Exp. Biol. Med. 2020, 168, 423–426. [Google Scholar] [CrossRef]
- Sikora, J.; Ouagazzal, A.-M. Synaptic Zinc: An Emerging Player in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4724. [Google Scholar] [CrossRef]
- Gao, H.; Dai, W.; Zhao, L.; Min, J.; Wang, F. The Role of Zinc and Zinc Homeostasis in Macrophage Function. J. Immunol. Res. 2018, 2018, 6872621. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.M.; O’Keeffe, G.W.; Long-Smith, C.M.; Wyatt, S.L.; Sullivan, A.M.; Toulouse, A.; Nolan, Y.M. Mitogen-activated protein kinase phosphatase (MKP)-1 as a neuroprotective agent: Promotion of the morphological development of midbrain dopaminergic neurons. Neuromol. Med. 2013, 15, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Halliday, G.M. Aspects of innate immunity and Parkinson’s disease. Front. Pharmacol. 2012, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Age at Exam, Mean ± SD, Years | Age at Onset, Mean ± SD, Years | Gender (Male:Female) | Mutations in the GBA Gene |
|---|---|---|---|---|
| GBA-PD, N = 5 | 53.5 ± 8.73 | 49.0 ± 10.89 | 3:2 | 4 L444P/N 1 N370S/N |
| GBA-carriers, N = 4 | 54.9 ± 8.9 | - | 2:2 | 3 L444P/N 1 N370S/N |
| Controls, N = 4 | 54.4 ± 9.5 | - | 2:2 | - |
| (GO ID] GO Terms | padjusted | DEGs |
|---|---|---|
| L444P/N GBA-PD vs. Controls | ||
| (GO:0097529) myeloid leukocyte migration | 6.93 × 10−9 | CCL3, CCL3L1, CCL4, CXCL2, CXCL5, DUSP1, IL6, PPBP |
| (GO:0002548) monocyte chemotaxis | 6.93 × 10−9 | CCL3, CCL3L1, CCL4, DUSP1, IL6 |
| (GO:1990869) cellular response to chemokine | 6.93 × 10−9 | CCL3, CCL3L1, CCL4, CXCL2, CXCL5, DUSP1, PPBP |
| (GO:0030593) neutrophil chemotaxis | 6.93 × 10−9 | CCL3, CCL3L1, CCL4, CXCL2, CXCL5, PPBP |
| L444P/N GBA-PD vs. L444P/N GBA carriers | ||
| (GO:0006882) cellular zinc ion homeostasis | 8.09 × 10−9 | MT1F, MT1L, MT1M, MT1X, SLC39A8 |
| (GO:0010273) detoxification of copper ion | 9.81 × 10−7 | MT1F, MT1L, MT1M, MT1X |
| (GO:0071276) cellular response to cadmium ion | 9.81 × 10−7 | MT1F, MT1L, MT1M, MT1X |
| (GO:0071280) cellular response to copper ion | 9.81 × 10−7 | MT1F, MT1L, MT1M, MT1X |
| (GO:0071294) cellular response to zinc ion | 9.81 × 10−7 | MT1F, MT1L, MT1M, MT1X |
| L444P/N GBA carriers vs. controls | ||
| (GO:0048731) system development | 0.001035 | ACOD1, IL31RA, KIAA0319, OSCAR, TBX3 |
| (GO:0002520) immune system development | 0.001035 | ACOD1, IL31RA, OSCAR |
| (GO:0002573) myeloid leukocyte differentiation | 0.001011 | IL31RA, OSCAR |
| (GO:0048692) negative regulation of axon extension involved in regeneration | 0.000615 | KIAA0319 |
| (GO:0048677) axon extension involved in regeneration | 0.000615 | KIAA0319 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usenko, T.; Bezrukova, A.; Basharova, K.; Panteleeva, A.; Nikolaev, M.; Kopytova, A.; Miliukhina, I.; Emelyanov, A.; Zakharova, E.; Pchelina, S. Comparative Transcriptome Analysis in Monocyte-Derived Macrophages of Asymptomatic GBA Mutation Carriers and Patients with GBA-Associated Parkinson’s Disease. Genes 2021, 12, 1545. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101545
Usenko T, Bezrukova A, Basharova K, Panteleeva A, Nikolaev M, Kopytova A, Miliukhina I, Emelyanov A, Zakharova E, Pchelina S. Comparative Transcriptome Analysis in Monocyte-Derived Macrophages of Asymptomatic GBA Mutation Carriers and Patients with GBA-Associated Parkinson’s Disease. Genes. 2021; 12(10):1545. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101545
Chicago/Turabian StyleUsenko, Tatiana, Anastasia Bezrukova, Katerina Basharova, Alexandra Panteleeva, Mikhail Nikolaev, Alena Kopytova, Irina Miliukhina, Anton Emelyanov, Ekaterina Zakharova, and Sofya Pchelina. 2021. "Comparative Transcriptome Analysis in Monocyte-Derived Macrophages of Asymptomatic GBA Mutation Carriers and Patients with GBA-Associated Parkinson’s Disease" Genes 12, no. 10: 1545. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101545