
The Genetic Landscape of Mitochondrial Diseases in Spain: A Nationwide Call

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source and Patient Selection Protocol
2.2. Selection of Genes
2.3. Estimation of the Number of Patients
2.4. Genetic Diagnoses
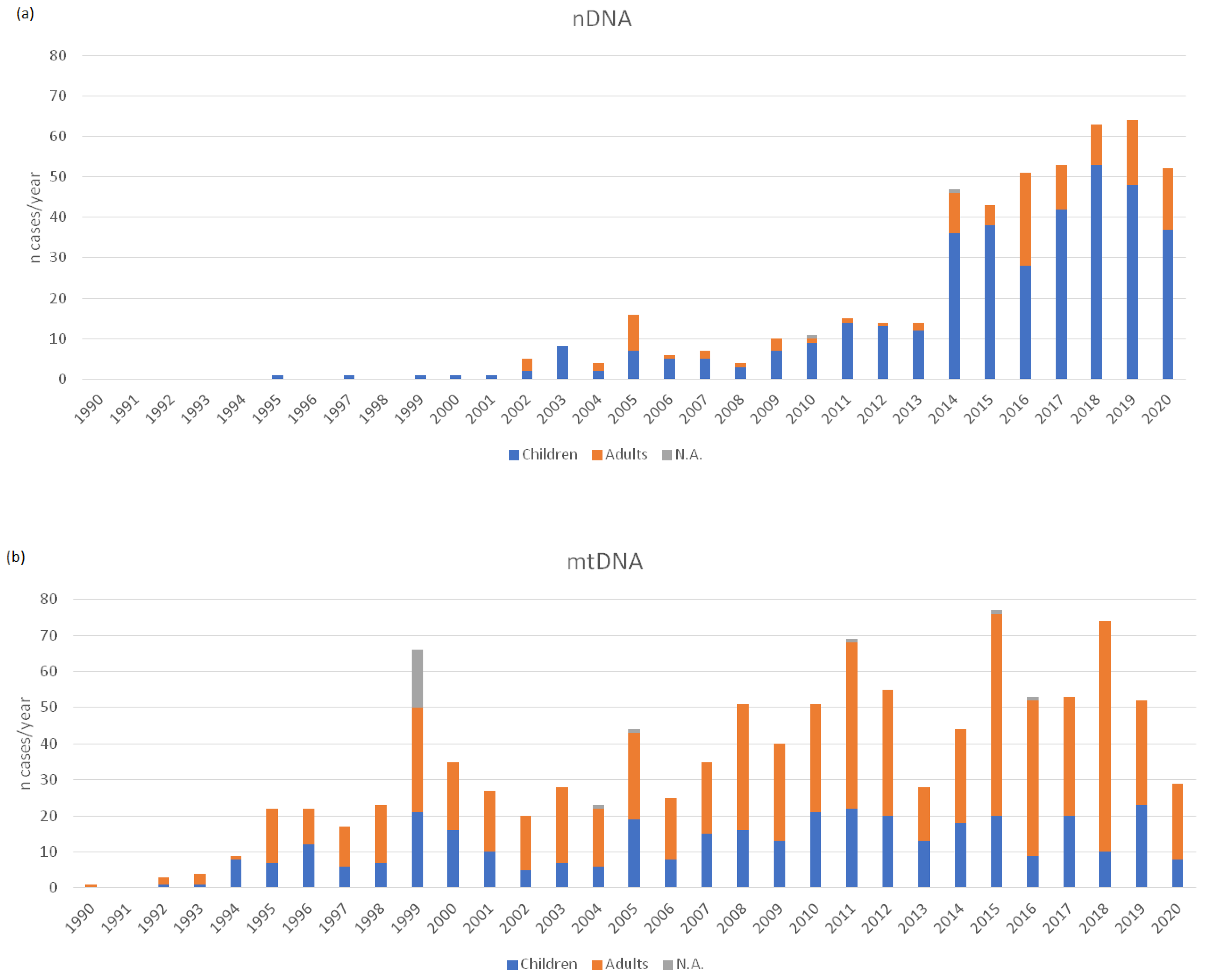
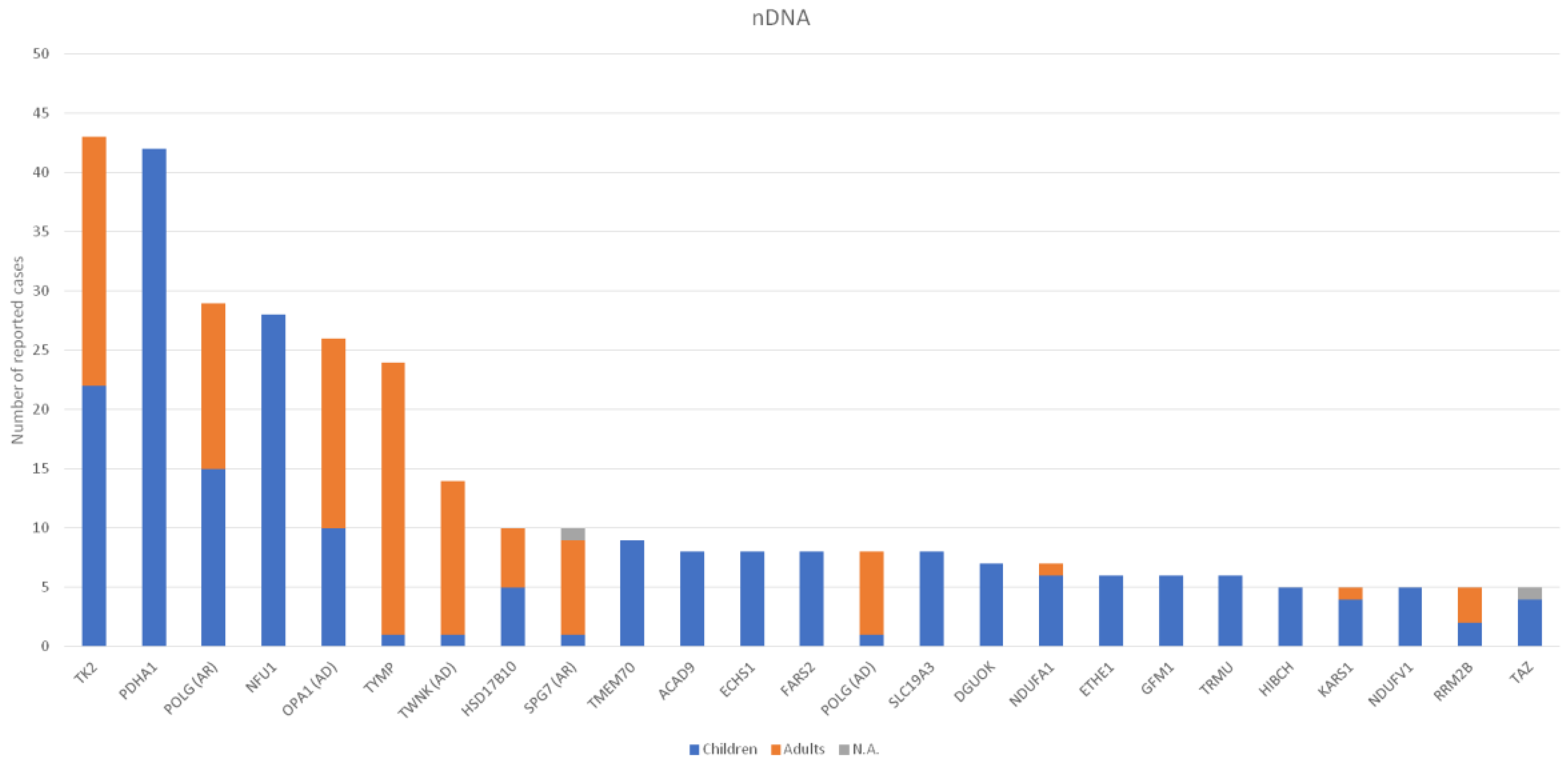
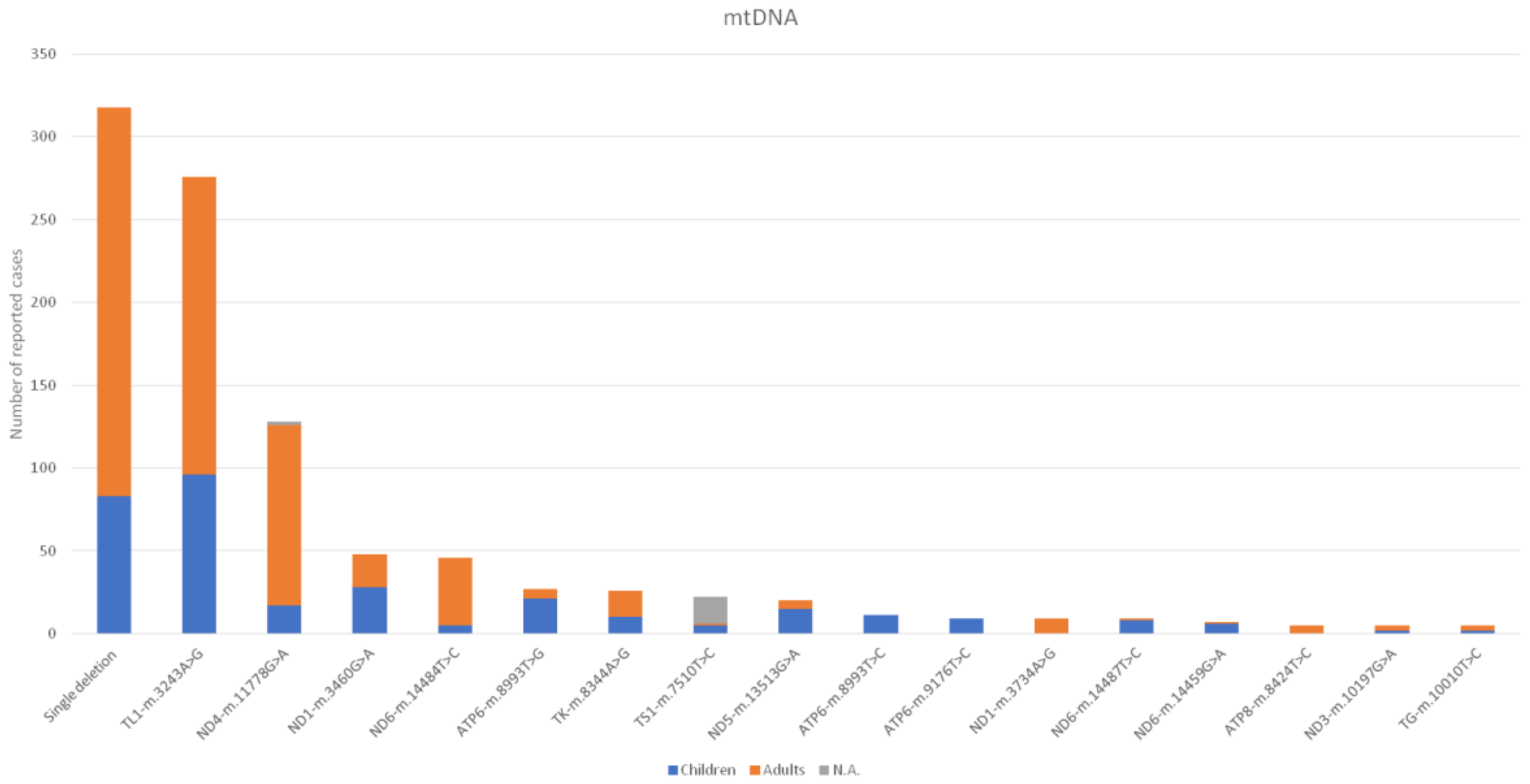
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. MITOSPAIN Working Group
References
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 20, 16080. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 26, 600079. [Google Scholar] [CrossRef]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.; King, M.D.; Rustin, P.; Mayne, P.D.; Brown, G.K.; Monavari, A.A.; Walsh, R.; Treacy, E.P. Mitochondrial cytopathies, phenotypic heterogeneity and a high incidence. Iran. Med. J. 2006, 99, 262–264. [Google Scholar]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Darin, N.; Oldfors, A.; Moslemi, A.R.; Holme, E.; Tulinius, M. The incidence of mitochondrial encephalomyopathies in childhood: Clinical features and morphological, biochemical, and DNA abnormalities. Ann. Neurol. 2001, 49, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Diogo, L.; Grazina, M.; Garcia, P.; Rebelo, O.; Veiga, M.A.; Cuevas, J.; Vilarinho, L.; de Almeida, I.T.; Oliveira, C.R. Pediatric mitochondrial respiratory chain disorders in the Centro region of Portugal. Pediatr. Neurol. 2009, 40, 351–356. [Google Scholar] [CrossRef]
- Castro-Gago, M.; Blanco-Barca, M.O.; Campos-González, Y.; Arenas-Barbero, J.; Pintos-Martínez, E.; Eirís-Puñal, J. Epidemiology of pediatric mitochondrial respiratory chain disorders in northwest Spain. Pediatr. Neurol. 2006, 3, 204–211. [Google Scholar] [CrossRef]
- Uusimaa, J.; Remes, A.M.; Rantala, H.; Vainionpää, L.; Herva, R.; Vuopala, K.; Nuutinen, M.; Majamaa, K.; Hassinen, I.E. Childhood encephalopathies and myopathies: A prospective study in a defined population to assess the frequency of mitochondrial disorders. Pediatrics 2000, 105, 598–603. [Google Scholar] [CrossRef]
- Uusimaa, J.; Moilanen, J.S.; Vainionpää, L.; Tapanainen, P.; Lindholm, P.; Nuutinen, M.; Löppönen, T.; Mäki-Torkko, E.; Rantala, H.; Majamaa, K. Prevalence, segregation, and phenotype of the mitochondrial DNA 3243A>G mutation in children. Ann. Neurol. 2007, 62, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Yatsuga, S.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Kakuma, T.; Koga, Y.; Taro Matsuoka for MELAS Study Group in Japan. MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim. Biophys. Acta 2012, 1820, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Ibayashi, K.; Fujino, Y.; Mimaki, M.; Fujimoto, K.; Matsuda, S.; Goto, Y.I. Estimation of the number of patients with mitochondrial diseases: A descriptive study using a nationwide database in Japan. J. Epidemiol. 2021, 28, JE20200577. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Gómez-Durán, A.; López-Pérez, M.J.; Montoya, J.; Ruiz-Pesini, E. Mitochondrial pharmacogenomics: Barcode for antibiotic therapy. Drug Discov. Today 2010, 15, 33–39. [Google Scholar] [CrossRef]
- Witters, P.; Saada, A.; Honzik, T.; Tesarova, M.; Kleinle, S.; Horvath, R.; Goldstein, A.; Morava, E. Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genet. Med. 2018, 20, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, D.R. Mitochondrial disorders: Prevalence, myths and advances. J. Inherit. Metab. Dis. 2004, 27, 349–362. [Google Scholar] [CrossRef]
- Lebon, S.; Chol, M.; Benit, P.; Mugnier, C.; Chretien, D.; Giurgea, I.; Kern, I.; Girardin, E.; Hertz-Pannier, L.; de Lonlay, P.; et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J. Med. Genet. 2003, 40, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population prevalence of the MELAS A3243G mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef]
- Clarke, C.; Xiao, R.; Place, E.; Zhang, Z.; Sondheimer, N.; Bennett, M.; Yudkoff, M.; Falk, M.J. Mitochondrial respiratory chain disease discrimination by retrospective cohort analysis of blood metabolites. Mol. Genet. Metab. 2013, 110, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martí, R.; López, L.C.; Hirano, M. Assessment of thymidine phosphorylase function: Measurement of plasma thymidine (and deoxyuridine) and thymidine phosphorylase activity. Methods Mol. Biol. 2012, 837, 121–133. [Google Scholar]
- Domínguez-González, C.; Hernández-Laín, A.; Rivas, E.; Hernández-Voth, A.; Sayas-Catalán, J.; Fernández-Torrón, R.; Fuiza-Luces, C.; García García, J.; Morís, G.; Olivé, M.; et al. Late-onset thymidine kinase 2 deficiency: A review of 18 cases. Orphanet J. Rare Dis. 2019, 14, 100. [Google Scholar] [CrossRef] [Green Version]
- Garone, C.; Tadesse, S.; Hirano, M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 2011, 134, 3326–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandebona, H.; Mitchell, P.; Manwaring, N.; Griffiths, K.; Gopinath, B.; Wang, J.J.; Sue, C.M. Prevalence of mitochondrial 1555A-->G mutation in adults of European descent. N. Engl. J. Med. 2009, 360, 642–644. [Google Scholar] [CrossRef]
- Bitner-Glindzicz, M.; Pembrey, M.; Duncan, A.; Heron, J.; Ring, S.M.; Hall, A.; Rahman, S. Prevalence of mitochondrial 1555A-->G mutation in European children. N. Engl. J. Med. 2009, 360, 640–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alston, C.L.; Stenton, S.L.; Hudson, G.; Prokisch, H.; Taylor, R.W. The genetics of mitochondrial disease: Dissecting mitochondrial pathology using multi-omic pipelines. J. Pathol. 2021, 254, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sainz, A.G.; Shadel, G.S. Mitochondrial DNA: Cellular genotoxic stress sentinel. Trends Biochem. Sci. 2021, 46, 812–821. [Google Scholar] [CrossRef]
- Martín-Navarro, A.; Gaudioso-Simón, A.; Álvarez-Jarreta, J.; Montoya, J.; Mayordomo, E.; Ruiz-Pesini, E. Machine learning classifier for identification of damaging missense mutations exclusive to human mitochondrial DNA-encoded polypeptides. BMC Bioinform. 2017, 18, 158. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Year | Paediatrics 1 | Adults 1 | Global 1 |
|---|---|---|---|
| 2014 | 6.15 (4.51–7.80) | 0.95 (0.64–1.27) | 1.93 (1.54–2.33) |
| 2015 | 6.61 (4.91–8.31) | 1.62 (1.21–2.03) | 2.56 (2.10–3.02) |
| 2016 | 4.21 (2.86–5.57) | 1.75 (1.33–2.18) | 2.22 (1.79–2.65) |
| 2017 | 7.05 (5.30–8.81) | 1.17 (0.82–1.51) | 2.28 (1.84–2.71) |
| 2018 | 7.16 (5.39–8.92) | 1.95 (1.51–2.40) | 2.94 (2.44–3.43) |
| 2019 | 8.07 (6.19–9.94) | 1.18 (0.84–1.52) | 2.47 (2.02–2.92) |
| 2020 | 5.11 (3.62–6.61) | 0.93 (0.63–1.24) | 1.71 (1.34–2.08) |
| 2014–2020 | 6.34 (5.71–6.97) | 1.36 (1.22–1.50) | 2.30 (2.14–2.47) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellusci, M.; Paredes-Fuentes, A.J.; Ruiz-Pesini, E.; Gómez, B.; MITOSPAIN Working Group; Martín, M.A.; Montoya, J.; Artuch, R. The Genetic Landscape of Mitochondrial Diseases in Spain: A Nationwide Call. Genes 2021, 12, 1590. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101590
Bellusci M, Paredes-Fuentes AJ, Ruiz-Pesini E, Gómez B, MITOSPAIN Working Group, Martín MA, Montoya J, Artuch R. The Genetic Landscape of Mitochondrial Diseases in Spain: A Nationwide Call. Genes. 2021; 12(10):1590. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101590
Chicago/Turabian StyleBellusci, Marcello, Abraham J Paredes-Fuentes, Eduardo Ruiz-Pesini, Beatriz Gómez, MITOSPAIN Working Group, Miguel A Martín, Julio Montoya, and Rafael Artuch. 2021. "The Genetic Landscape of Mitochondrial Diseases in Spain: A Nationwide Call" Genes 12, no. 10: 1590. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101590