
Disruptive NADSYN1 Variants Implicated in Congenital Vertebral Malformations

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Clinical Evaluation
2.2. Exome Sequencing and Variant Interpretation
2.3. Site-Directed Mutagenesis Plasmid Construction
2.4. Plasmid Transfection and Western Blots
2.5. Expression and Purification of NADSYN1 Protein in Mammalian COS-7 cells
2.6. Enzymatic Assays of NADSYN1 Protein
3. Results
3.1. Identification and Prioritization of NADSYN1 Variants Implicated in DISCO Cohort
3.2. Phenotypic Characteristics of Individuals with NADSYN1 Variants
3.3. Distribution of the NADSYN1 Variants on the Protein
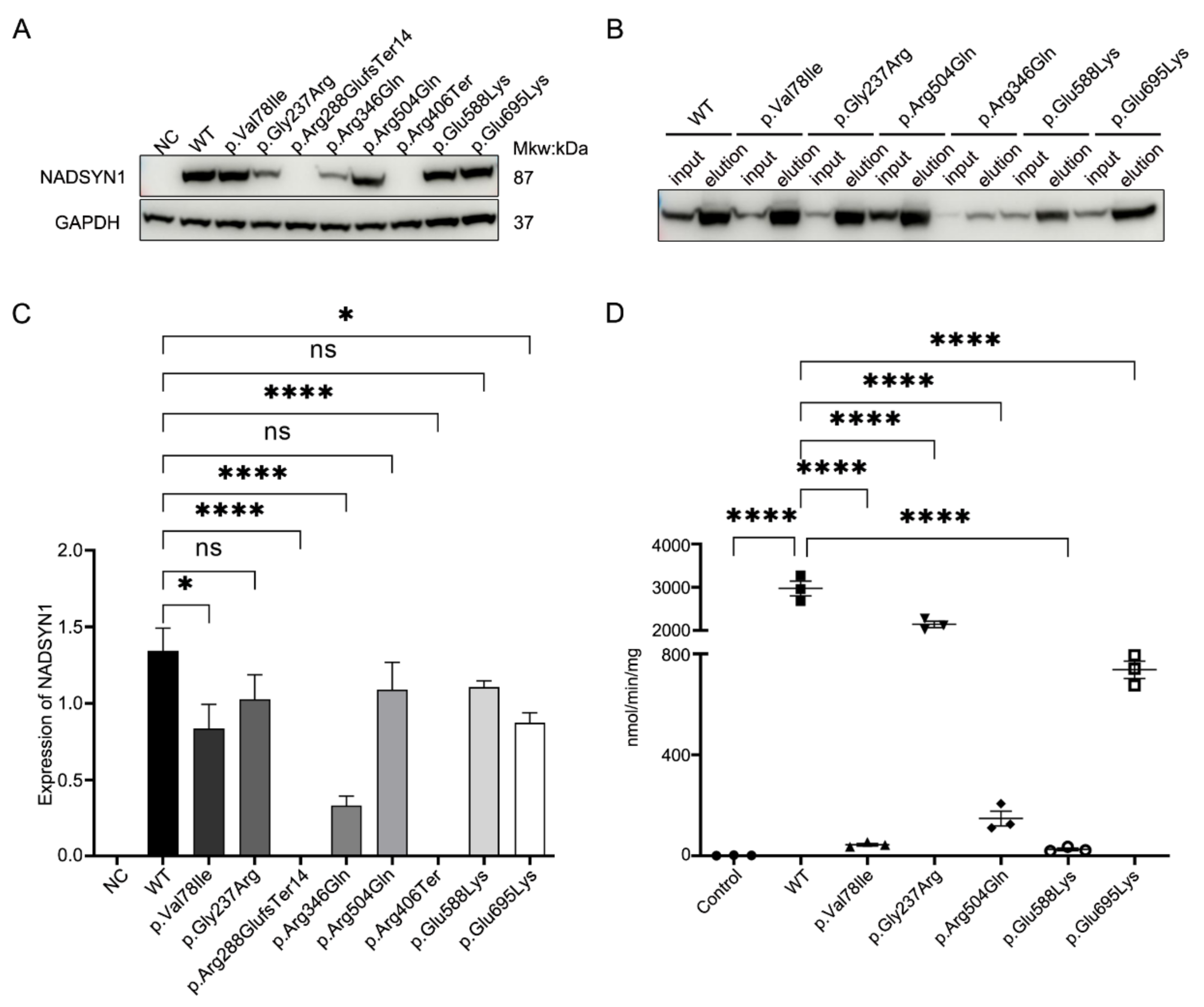
3.4. Functional Assessment of the NADSYN1 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wynne-Davies, R. Congenital vertebral anomalies: Aetiology and relationship to spina bifida cystica. J. Med. Genet. 1975, 12, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Giampietro, P.F.; Dunwoodie, S.L.; Kusumi, K.; Pourquie, O.; Tassy, O.; Offiah, A.C.; Cornier, A.S.; Alman, B.A.; Blank, R.D.; Raggio, C.L.; et al. Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans. Ann. N. Y. Acad. Sci. 2009, 1151, 38–67. [Google Scholar] [CrossRef]
- Furdock, R.; Brouillet, K.; Luhmann, S.J. Organ System Anomalies Associated with Congenital Scoliosis: A Retrospective Study of 305 Patients. J. Pediatr. Orthop. 2019, 39, e190–e194. [Google Scholar] [CrossRef]
- Pahys, J.M.; Guille, J.T. What’s New in Congenital Scoliosis? J. Pediatr. Orthop. 2018, 38, e172–e179. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, S.; Jolly, A.; Wang, L.; Pan, H.; Yuan, J.; Chen, S.; Koch, A.; Ma, C.; Tian, W.; et al. Perturbations of genes essential for Mullerian duct and Wolffian duct development in Mayer-Rokitansky-Kuster-Hauser syndrome. Am. J. Hum. Genet. 2021, 108, 337–345. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Chen, W.; Li, W.; Wang, S.; Wang, L.; Zhao, Y.; Lin, M.; Ye, Y.; Lin, J.; et al. Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS). J. Med. Genet. 2021, 58, 41–47. [Google Scholar] [CrossRef]
- Wu, N.; Ming, X.; Xiao, J.; Wu, Z.; Chen, X.; Shinawi, M.; Shen, Y.; Yu, G.; Liu, J.; Xie, H.; et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med. 2015, 372, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, D.B.; Chapman, G.; Smith, A.J.; Mattar, M.Z.; Major, J.A.; O’Reilly, V.C.; Saga, Y.; Zackai, E.H.; Dormans, J.P.; Alman, B.A.; et al. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell 2012, 149, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, N.; Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) Study; Yang, N.; Takeda, K.; Chen, W.; Li, W.; Du, R.; Liu, S.; Zhou, Y.; et al. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: Further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet. Med. 2019, 21, 1548–1558. [Google Scholar] [CrossRef]
- Chen, W.; Lin, J.; Wang, L.; Li, X.; Zhao, S.; Liu, J.; Akdemir, Z.C.; Zhao, Y.; Du, R.; Ye, Y.; et al. TBX6 missense variants expand the mutational spectrum in a non-Mendelian inheritance disease. Hum. Mutat. 2020, 41, 182–195. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, A.; Kulikova, V.; Ziegler, M. The human NAD metabolome: Functions, metabolism and compartmentalization. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Szot, J.O.; Campagnolo, C.; Cao, Y.; Iyer, K.R.; Cuny, H.; Drysdale, T.; Flores-Daboub, J.A.; Bi, W.; Westerfield, L.; Liu, P.; et al. Bi-allelic Mutations in NADSYN1 Cause Multiple Organ Defects and Expand the Genotypic Spectrum of Congenital NAD Deficiency Disorders. Am. J. Hum. Genet. 2020, 106, 129–136. [Google Scholar] [CrossRef]
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 377, 544–552. [Google Scholar] [CrossRef]
- Cuny, H.; Rapadas, M.; Gereis, J.; Martin, E.; Kirk, R.B.; Shi, H.; Dunwoodie, S.L. NAD deficiency due to environmental factors or gene-environment interactions causes congenital malformations and miscarriage in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 3738–3747. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Chuenchor, W.; Doukov, T.I.; Chang, K.T.; Resto, M.; Yun, C.S.; Gerratana, B. Different ways to transport ammonia in human and Mycobacterium tuberculosis NAD(+) synthetases. Nat. Commun. 2020, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, N.; Yamada, K.; Terashima, M.; Osago, H.; Shimoyama, M.; Tsuchiya, M. Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J. Biol. Chem. 2003, 278, 10914–10921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massiere, F.; Badet-Denisot, M.A. The mechanism of glutamine-dependent amidotransferases. Cell Mol. Life Sci. 1998, 54, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, S.; Zhang, Y.; Wang, S.; Shao, J.; Liu, B.; Li, Y.; Yan, Z.; Niu, Y.; Li, X.; et al. Mutational burden and potential oligogenic model of TBX6-mediated genes in congenital scoliosis. Mol. Genet. Genom. Med. 2020, 8, e1453. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, S.; Cai, S.; Zhang, Y.; Wang, L.; Niu, Y.; Li, X.; Hu, J.; Chen, J.; Wang, S.; et al. The mutational burden and oligogenic inheritance in Klippel-Feil syndrome. BMC Musculoskelet Disord. 2020, 21, 220. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Pan, H.; Luo, G.; Wang, P.; Xie, Z.; Hua, K.; Luo, X.; Huang, X.; Liu, Q.; Sun, L.; et al. Clinical characteristics of 1055 Chinese patients with Mayer-Rokitansky-Kuster-Hauser syndrome: A nationwide multicentric study. Fertil. Steril. 2021, 116, 558–565. [Google Scholar] [CrossRef]

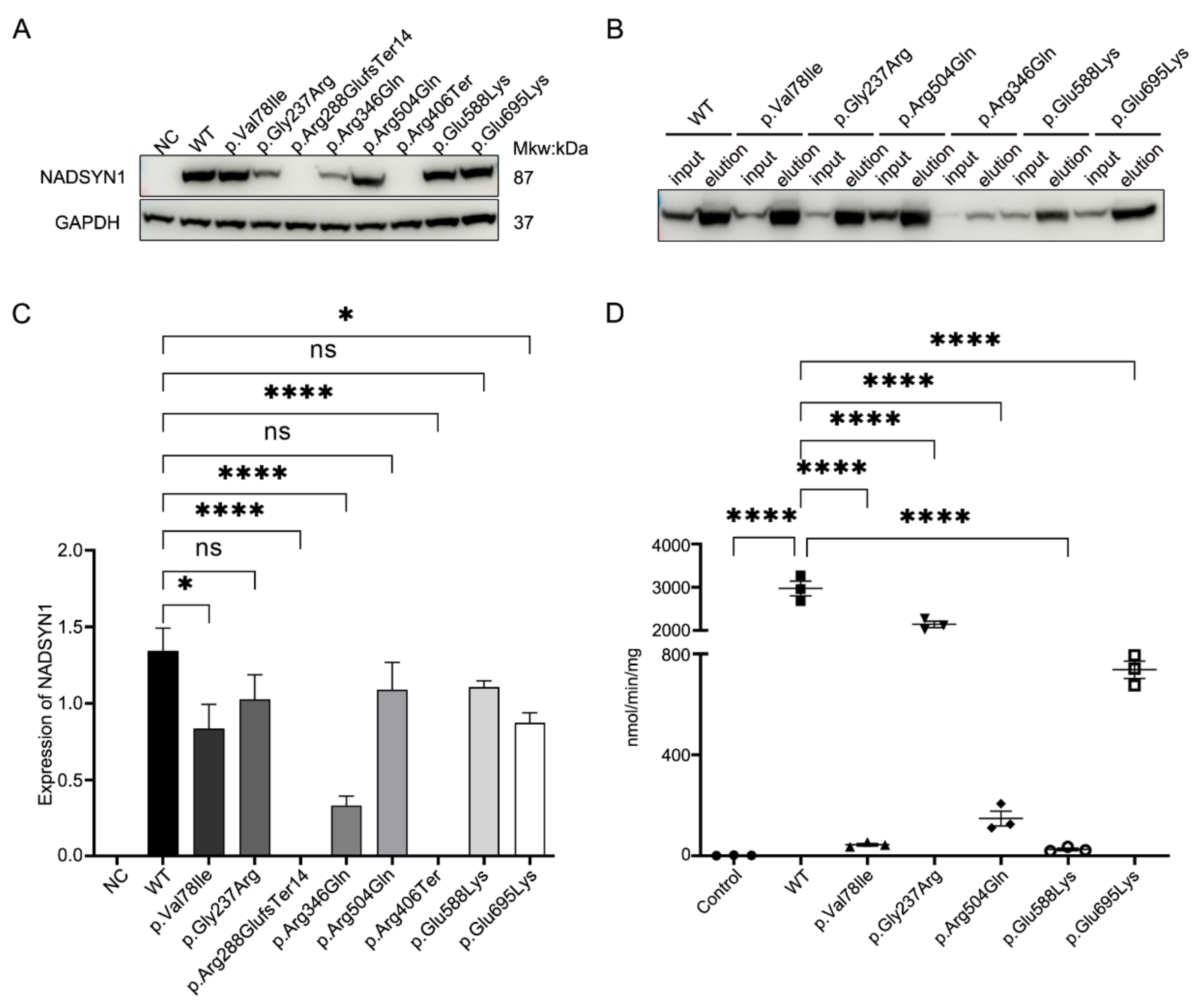

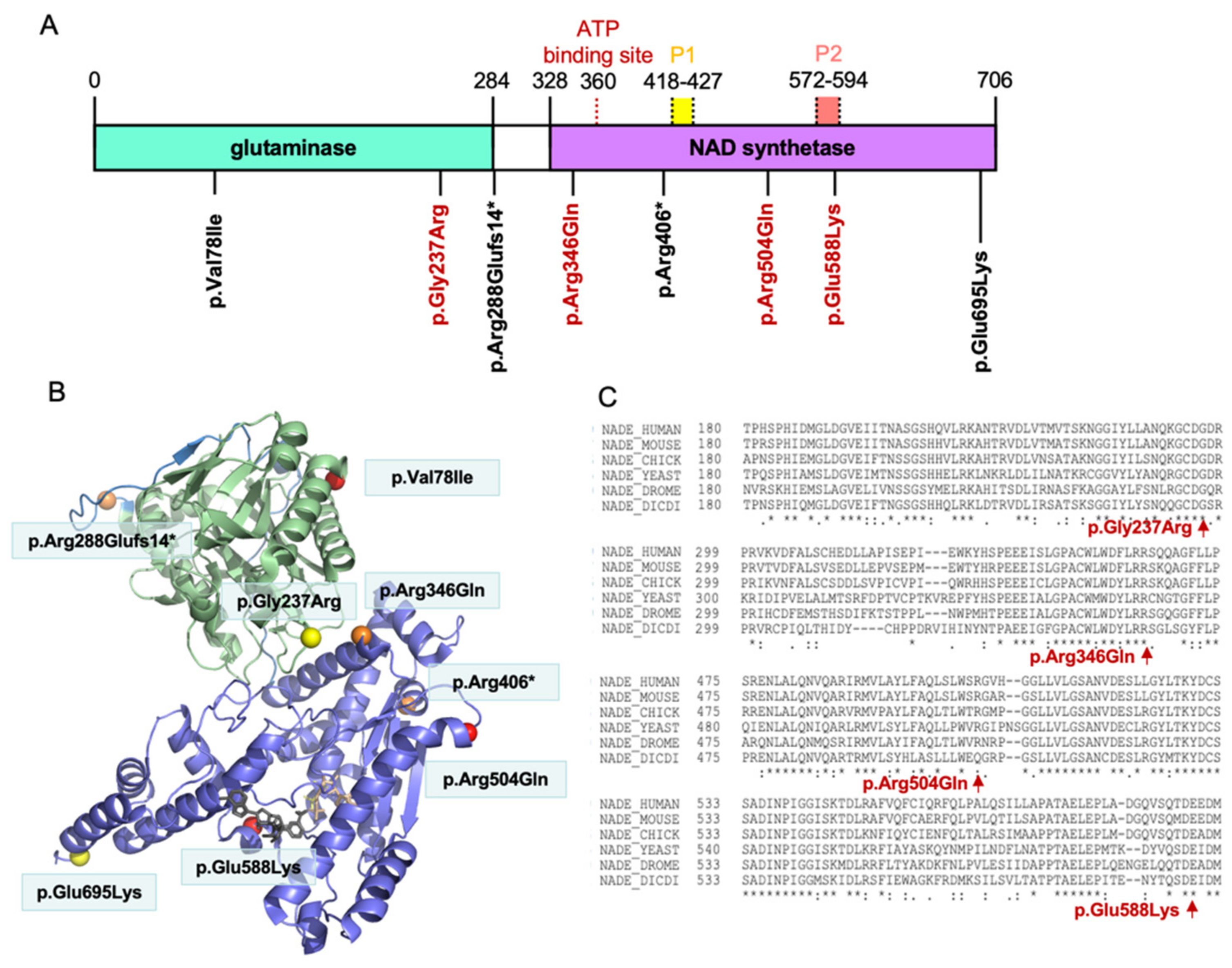{kind=link}
{kind=link}
| Patient Number | Variant | Zygosity | Sex | Age at Admission | Vertebral Deformities | Other Malformations |
|---|---|---|---|---|---|---|
| SCO2003P0298 | c.232G > A(p.Val78Ile) | Het | F | 12 yo | T3, T5, T6, T7, T8 vertebral malformation and unsegmentation, T10–T11 unsegmentation | Diastematomyelia, multiple hepatic polycysts, maxillary sinusitis |
| SCO2003P1273 | c.709G > A(p.Gly237Arg) | Het | M | 13 yo | T2–L3 vertebral malformation and unsegmentation | Diastematomyelia, spina bifida, mucocele |
| SCO2003P0164 | c.861delT(p.Arg288GlufsTer14) | Het | M | 11 yo | T6 butterfly vertebra, T7 hemivertebra, T5–T9 fused vertebral plate; T7 rib absent, T9–T10 fused rib | Renal aplasia (left) |
| SCO2003P0240 | c.1037G > A(p.Arg346Gln) | Het | M | 6 yo | T10, T11, T12 hemivertebrae | Bicuspid aortic valve |
| SCO2003P1980 | c.1216C > T(p.Arg406Ter) | Het | F | 1 yo | T6–T10 unsegmentation | Mitral insufficiency |
| SCO2003P0096 | c.1511G > A(p.Arg504Gln) | Het | F | 9 yo | L2 wedge-shape vertebra, L3 wedge-shape vertebra, L1–L4 unsegmentation | Tethered cord, multiple hepatic polycysts, splenomegaly |
| SCO2003P0286 | c.1762G > A(p.Glu588Lys) | Het | F | 1 yo | T3–T8 vertebral malformation and unsegmentation | Renal aplasia (left), polydactyly (left), dilatation of central canal of spinal cord |
| SCO2003P0106 | c.2083G > A(p.Glu695Lys) | Het | M | 10 yo | T7, T8, T9 butterfly vertebrae, T6–T10 unsegmentation | Diastematomyelia, mitral valve prolapse |
| SCO2003P0213 | c.2083G > A(p.Glu695Lys) | Het | M | 22 yo | T8–T9 unsegmentation, T9–T10 rib bifurcation | Diastematomyelia, tethered cord |
| Patient Number | cDNA Change | Protein Change | gnomAD * Allele Frequency | SIFT Prediction Score | Polyphen-2 | GERP++ | CADD-PHRED Score |
|---|---|---|---|---|---|---|---|
| SCO2003P0298 | 232G > A | Val78Ile | 0 | 0.5 | 0.013 | 4.35 | 18.37 |
| SCO2003P1273 | 709G > A | Gly237Arg | 0 | 0 | 0.974 | 4.64 | 23.4 |
| SCO2003P0164 | 861del | Arg288GlufsTer14 | 0 | NA | NA | NA | NA |
| SCO2003P0240 | 1037G > A | Arg346Gln | 0 | 0.01 | 0.994 | 5.11 | 29.6 |
| SCO2003P1980 | 1216C > T | Arg406Ter | 0.0000083 | NA | NA | 3.05 | 42 |
| SCO2003P0096 | 1511G > A | Arg504Gln | 0.00013 | 0.06 | 0.378 | 3.98 | 21.1 |
| SCO2003P0286 | 1762G > A | Glu588Lys | 0 | 0 | 1 | 4.83 | 20.6 |
| SCO2003P0106/SCO2003P0213 | 2083G > A | Glu695Lys | 0.00022 | 0.21 | 0.01 | 2.99 | 11.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Zhao, L.; Zhao, S.; Li, S.; Zhao, Z.; Chen, Z.; Zheng, Z.; Shao, J.; Niu, Y.; Li, X.; et al. Disruptive NADSYN1 Variants Implicated in Congenital Vertebral Malformations. Genes 2021, 12, 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101615
Lin J, Zhao L, Zhao S, Li S, Zhao Z, Chen Z, Zheng Z, Shao J, Niu Y, Li X, et al. Disruptive NADSYN1 Variants Implicated in Congenital Vertebral Malformations. Genes. 2021; 12(10):1615. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101615
Chicago/Turabian StyleLin, Jiachen, Lina Zhao, Sen Zhao, Shengjie Li, Zhengye Zhao, Zefu Chen, Zhifa Zheng, Jiashen Shao, Yuchen Niu, Xiaoxin Li, and et al. 2021. "Disruptive NADSYN1 Variants Implicated in Congenital Vertebral Malformations" Genes 12, no. 10: 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101615