
A KCNQ4 c.546C>G Genetic Variant Associated with Late Onset Non-Syndromic Hearing Loss in a Taiwanese Population

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Participants
2.3. Pure Tone Audiometry (PTA)
2.4. Phenome-Wide Association Studies (PheWAS)
2.5. Genetic Analysis
2.6. Statistical Analysis
3. Results
3.1. Genetic Variants Associated with Hearing Loss
3.2. Patient Characteristics and Audiograms
3.3. PheWAS of KCNQ4 c.546C>G Variant and Clinical Diagnosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stevens, G.; Flaxman, S.; Brunskill, E.; Mascarenhas, M.; Mathers, C.D.; Finucane, M. Global and regional hearing impairment prevalence: An analysis of 42 studies in 29 countries. Eur. J. Public Health 2013, 23, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Wake, M.; Tobin, S.; Cone-Wesson, B.; Dahl, H.H.; Gillam, L.; McCormick, L.; Poulakis, Z.; Rickards, F.W.; Saunders, K.; Ukoumunne, O.C.; et al. Slight/mild sensorineural hearing loss in children. Pediatrics 2006, 118, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Feder, K.P.; Michaud, D.; McNamee, J.; Fitzpatrick, E.; Ramage-Morin, P.; Beauregard, Y. Prevalence of Hearing Loss among a Representative Sample of Canadian Children and Adolescents, 3 to 19 Years of Age. Ear Heart 2017, 38, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Homepage, H.H.L. Welcome to the Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 21 October 2021).
- Smith, R.J.; Bale, J.F., Jr.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Finsterer, J.; Fellinger, J. Nuclear and mitochondrial genes mutated in nonsyndromic impaired hearing. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 621–647. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Smith, R.J.H.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Holt, J.R.; Stauffer, E.A.; Abraham, D.; Geleoc, G.S. Dominant-negative inhibition of M-like potassium conductances in hair cells of the mouse inner ear. J. Neurosci. 2007, 27, 8940–8951. [Google Scholar] [CrossRef] [Green Version]
- Kharkovets, T.; Hardelin, J.P.; Safieddine, S.; Schweizer, M.; El-Amraoui, A.; Petit, C.; Jentsch, T.J. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4333–4338. [Google Scholar] [CrossRef] [Green Version]
- Carignano, C.; Barila, E.P.; Rias, E.I.; Dionisio, L.; Aztiria, E.; Spitzmaul, G. Inner Hair Cell and Neuron Degeneration Contribute to Hearing Loss in a DFNA2-Like Mouse Model. Neuroscience 2019, 410, 202–216. [Google Scholar] [CrossRef]
- Cooper, D.N.; Stenson, P.D.; Phillips, A.D.; Evans, K.; Heywood, S.; Hayden, M.J.; Chapman, M.M.; Mort, M.E.; Azevedo, L.; Millar, D.S. The Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=KCNQ4 (accessed on 21 October 2021).
- ClinVar. KCNQ4 and Pathogenic. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/?term=KCNQ4+and+pathogenic (accessed on 21 October 2021).
- Ramzan, M.; Idrees, H.; Mujtaba, G.; Sobreira, N.; Witmer, P.D.; Naz, S. Bi-allelic Pro291Leu variant in KCNQ4 leads to early onset non-syndromic hearing loss. Gene 2019, 705, 109–112. [Google Scholar] [CrossRef]
- Kim, H.J.; Lv, P.; Sihn, C.R.; Yamoah, E.N. Cellular and molecular mechanisms of autosomal dominant form of progressive hearing loss, DFNA2. J. Biol. Chem. 2011, 286, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Su, C.C.; Yang, J.J.; Shieh, J.C.; Su, M.C.; Li, S.Y. Identification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from Taiwan. Audiol. Neurootol. 2007, 12, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Lin, H.; Koh, Y.I.; Ryu, K.; Lee, J.S.; Rim, J.H.; Choi, H.J.; Lee, H.J.; Kim, H.Y.; Yu, S.; et al. Rare KCNQ4 variants found in public databases underlie impaired channel activity that may contribute to hearing impairment. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.C.; Li, S.Y.; Yang, J.J.; Su, M.C.; Lin, M.J. Studies of the effect of ionomycin on the KCNQ4 channel expressed in Xenopus oocytes. Biochem. Biophys. Res. Commun. 2006, 348, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.Y.; Yang, J.H.; Yeh, E.C.; Tsai, M.F.; Kao, H.J.; Lo, C.Z.; Chang, L.P.; Lin, W.J.; Hsieh, F.J.; Belsare, S.; et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom. Med. 2021, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Kubisch, C.; Schroeder, B.C.; Friedrich, T.; Lutjohann, B.; El-Amraoui, A.; Marlin, S.; Petit, C.; Jentsch, T.J. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999, 96, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Coucke, P.J.; Van Hauwe, P.; Kelley, P.M.; Kunst, H.; Schatteman, I.; Van Velzen, D.; Meyers, J.; Ensink, R.J.; Verstreken, M.; Declau, F.; et al. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum. Mol. Genet. 1999, 8, 1321–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto Pinheiro, B.; Vona, B.; Lowenheim, H.; Ruttiger, L.; Knipper, M.; Adel, Y. Age-related hearing loss pertaining to potassium ion channels in the cochlea and auditory pathway. Pflugers Arch. 2021, 473, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ding, E.; Sheng, R.; Cheng, J.; Cai, W.; Guo, J.; Wang, N.; Zhang, H.; Zhu, B. Genetic variation in KCNQ4 gene is associated with susceptibility to noise-induced hearing loss in a Chinese population. Environ. Toxicol. Pharmacol. 2018, 63, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Chadha, P.S.; Zunke, F.; Zhu, H.L.; Davis, A.J.; Jepps, T.A.; Olesen, S.P.; Cole, W.C.; Moffatt, J.D.; Greenwood, I.A. Reduced KCNQ4-encoded voltage-dependent potassium channel activity underlies impaired beta-adrenoceptor-mediated relaxation of renal arteries in hypertension. Hypertension 2012, 59, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Xu, Y.; An, M.; Zeng, Q. The risk factors for diabetic peripheral neuropathy: A meta-analysis. PLoS ONE 2019, 14, e0212574. [Google Scholar] [CrossRef]
- Kent, K.C.; Zwolak, R.M.; Egorova, N.N.; Riles, T.S.; Manganaro, A.; Moskowitz, A.J.; Gelijns, A.C.; Greco, G. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J. Vasc. Surg. 2010, 52, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Lederle, F.A.; Johnson, G.R.; Wilson, S.E.; Chute, E.P.; Hye, R.J.; Makaroun, M.S.; Barone, G.W.; Bandyk, D.; Moneta, G.L.; Makhoul, R.G. The aneurysm detection and management study screening program: Validation cohort and final results. Aneurysm Detection and Management Veterans Affairs Cooperative Study Investigators. Arch. Intern. Med. 2000, 160, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, H.G.; Wolfson, S.K., Jr.; Sutton-Tyrrell, K.; Kuller, L.H.; O’Leary, D. Risk factors for abdominal aortic aneurysms in older adults enrolled in The Cardiovascular Health Study. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 963–970. [Google Scholar] [CrossRef]
- Marres, H.; van Ewijk, M.; Huygen, P.; Kunst, H.; van Camp, G.; Coucke, P.; Willems, P.; Cremers, C. Inherited nonsyndromic hearing loss. An audiovestibular study in a large family with autosomal dominant progressive hearing loss related to DFNA2. Arch. Otolaryngol. Head Neck Surg. 1997, 123, 573–577. [Google Scholar] [CrossRef] [Green Version]
- De Leenheer, E.M.; Huygen, P.L.; Coucke, P.J.; Admiraal, R.J.; van Camp, G.; Cremers, C.W. Longitudinal and cross-sectional phenotype analysis in a new, large Dutch DFNA2/KCNQ4 family. Ann. Otol. Rhinol. Laryngol. 2002, 111, 267–274. [Google Scholar] [CrossRef]
- Lin, F.R.; Ferrucci, L. Hearing loss and falls among older adults in the United States. Arch. Intern. Med. 2012, 172, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Fornage, M.; Doris, P.A. Single-nucleotide polymorphism genotyping for disease association studies. Methods Mol. Med. 2005, 108, 159–172. [Google Scholar] [PubMed]



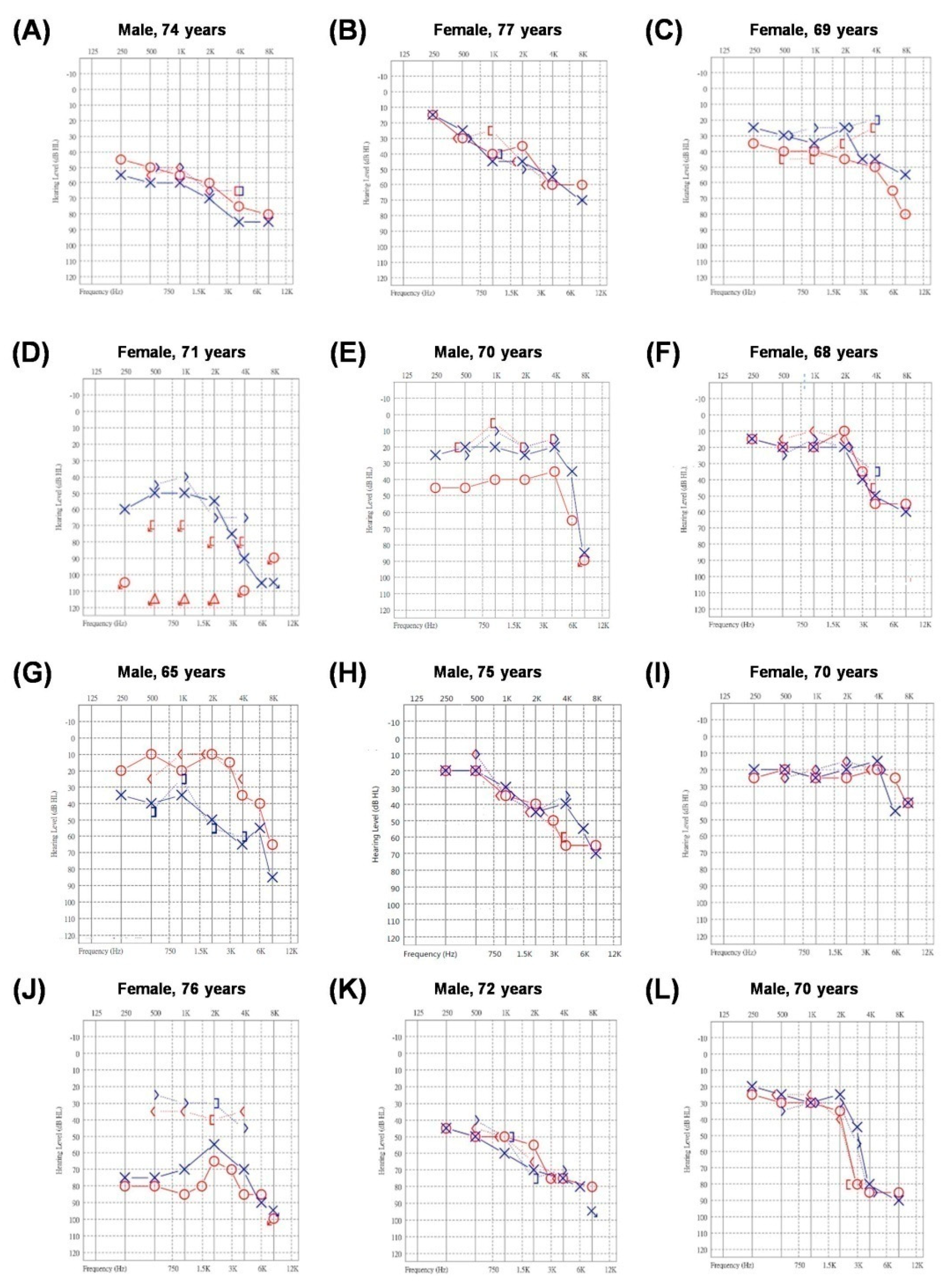{kind=link}
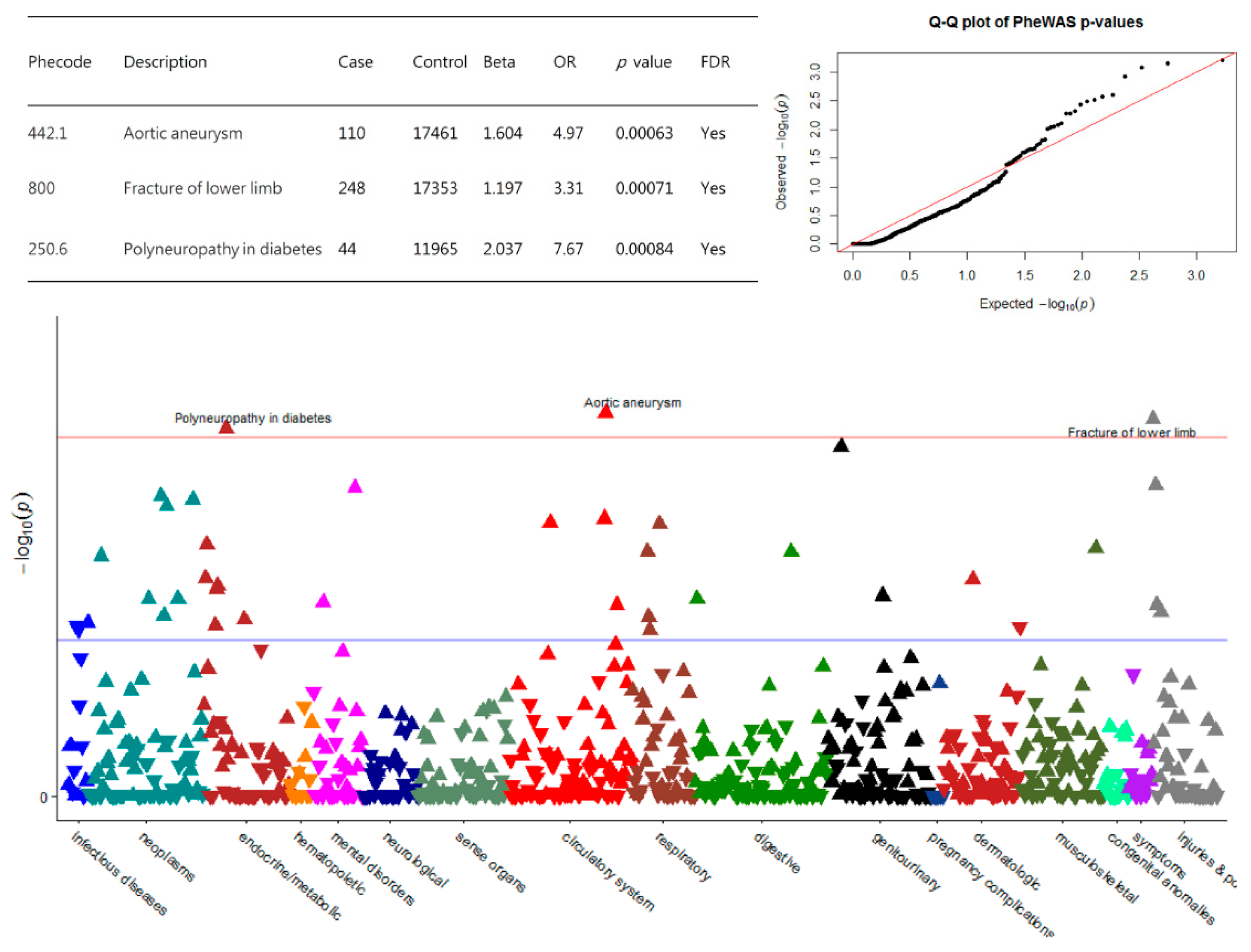{kind=link}
| CHR | SNP | Gene | Locus | Homozygote Genotype/Homoplasmic mtDNA Mutation | Heterozygote Genotype/Heteroplasmic mtDNA Mutation | MAF in TCVGH | MAF in GnomAD/HGDP-CEPH-db Supplement 1 (Population: East Asian) | Phenotype & Inheritance Mode |
|---|---|---|---|---|---|---|---|---|
| 1 | rs80358273 | KCNQ4 | DFNA2A | 1 | 398 | 0.61% | 0.00% | Nonsyndromic hearing loss, AD |
| c.546C>G | ||||||||
| 4 | rs387906930 | WFS1 | DFNA6 | 0 | 79 | 0.12% | 0.00% | Wolfram-like syndrome, AD |
| c.2051C>T | ||||||||
| 7 | rs111033313 | SLC26A4 | DFNB4 | 0 | 516 | 0.78% | 0.33% | Pendred syndrome, AR |
| c.919-2A>G | Pendred | |||||||
| 7 | rs121908362 | SLC26A4 | DFNB4 | 0 | 59 | 0.09% | 0.22% | Pendred syndrome, AR |
| c.2168A>G | Pendred | |||||||
| 9 | rs745750156 | CEP78 | 0 | 59 | 0.09% | 0.00% | Cone-rod dystrophy and hearing loss 1, AR | |
| c.1251+5G>A | ||||||||
| 13 | rs111033204 | GJB2 | DFNB1A | 0 | 67 | 0.10% | 0.00% | Nonsyndromic hearing loss, AR |
| c.299_300del | ||||||||
| 13 | rs80338943 | GJB2 | DFNB1A | 2 | 388 | 0.59% | 2.70% | Nonsyndromic hearing loss, AR |
| c.235del | ||||||||
| MT | rs121434453 | MT-TE | 37 | 0 | 0.12% | None | Diabetes-deafness syndrome | |
| m.14709T>C | ||||||||
| MT | rs267606617 | MT-RNR1 | 12rRNA | 44 | 0 | 0.13% | None | Aminoglycoside-induced deafness |
| m.1555A>G | ||||||||
| MT | rs267606618 | MT-RNR1 | 12rRNA | 72 | 0 | 0.22% | None | Aminoglycoside-induced deafness |
| m.1095T>C | ||||||||
| MT | rs28358569 | MT-RNR1 | 12rRNA | 1476 | 0 | 4.49% | 0.40% | Aminoglycoside-induced deafness |
| m.827A>G |
| Variables a | Case Group | Control Group | p Value b | ||
|---|---|---|---|---|---|
| (SNHL/MHL n = 236) | (non SNHL/MHL n = 107) | ||||
| n | % | n | % | ||
| Age (years) | <0.0001 | ||||
| 18–64 | 41 | 17.37 | 65 | 60.75 | |
| ≥65 | 195 | 82.63 | 42 | 39.25 | |
| Gender | 0.003 | ||||
| Male | 140 | 59.3 | 45 | 42.06 | |
| Female | 96 | 40.7 | 62 | 57.9 | |
| Hearing status | <0.0001 | ||||
| Normal <20 dB | 0 | 0.0 | 59 | 55.1 | |
| Mild 20–40 dB | 70 | 29.7 | 48 | 44.9 | |
| Moderate 40–70 dB | 119 | 50.4 | 0 | 0.0 | |
| Severe 70–95 dB | 30 | 12.7 | 0 | 0.0 | |
| Profound >95 dB | 17 | 7.2 | 0 | 0.0 | |
| SNPs | |||||
| KCNQ4 c.546C>G | 12 | 5.1 | 0 | 0.0 | 0.02 |
| WFS1 c.2051C>T | 0 | 0.0 | 0 | 0.0 | |
| SLC26A4 c.919-2A>G | 3 | 1.3 | 3 | 2.8 | 0.38 |
| SLC26A4 c.2168A>G | 0 | 0.0 | 0 | 0.0 | |
| CEP78 c.1251+5G>A | 1 | 0.4 | 1 | 0.9 | 0.53 |
| GJB2 c.299_300del | 0 | 0.0 | 0 | 0.0 | |
| GJB2 c.235del | 4 | 1.7 | 0 | 0.0 | 0.31 |
| Comorbidities | |||||
| Hyperlipidemia | 162 | 68.6 | 73 | 68.2 | 0.94 |
| Hypertension | 183 | 77.5 | 65 | 60.8 | 0.001 |
| Diabetes mellitus | 159 | 67.4 | 67 | 62.6 | 0.39 |
| Coronary artery disease | 93 | 39.4 | 36 | 33.6 | 0.31 |
| Chronic Kidney Disease | 143 | 60.6 | 48 | 44.9 | 0.007 |
| Variables a | Carriers (n = 12) | Non-Carriers (n = 224) | ||
|---|---|---|---|---|
| n | % | n | % | |
| Age (years) | ||||
| 18–64 | 0 | 0 | 41 | 18.3 |
| ≥65 | 12 | 100 | 183 | 81.7 |
| Gender | ||||
| Male | 6 | 50.0 | 90 | 40.18 |
| Female | 6 | 50.0 | 134 | 59.8 |
| Hearing status | ||||
| Normal <20 dB | 0 | 0.0 | 0 | 0.0 |
| Mild 20–40 dB | 2 | 16.7 | 68 | 30.4 |
| Moderate 40–70 dB | 8 | 66.7 | 111 | 49.6 |
| Severe 70–95 dB | 1 | 8.3 | 29 | 13.0 |
| Profound >95 dB | 1 | 8.3 | 16 | 7.1 |
| Comorbidities | ||||
| Hyperlipidemia | 8 | 66.7 | 154 | 68.8 |
| Hypertension | 9 | 75.0 | 174 | 77.7 |
| Diabetes mellitus | 8 | 66.7 | 151 | 67.4 |
| Coronary artery disease | 5 | 41.7 | 88 | 39.3 |
| Chronic Kidney Disease | 10 | 83.3 | 133 | 59.4 |
| Variable | OR | 95% CI | p Value a | |
|---|---|---|---|---|
| Hearing status (ref = Mild 20–39 dB) | ||||
| Moderate 40–69 dB | 4.20 | 0.49 | 36.26 | 0.43 |
| Severe 70–95 dB | 2.67 | 0.15 | 47.30 | 0.99 |
| Profound >95 dB | 4.80 | 0.26 | 90.30 | 0.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yen, T.-T.; Chen, I.-C.; Hua, M.-W.; Wei, C.-Y.; Shih, K.-H.; Li, J.-L.; Lin, C.-H.; Hsiao, T.-H.; Chen, Y.-M.; Jiang, R.-S. A KCNQ4 c.546C>G Genetic Variant Associated with Late Onset Non-Syndromic Hearing Loss in a Taiwanese Population. Genes 2021, 12, 1711. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111711
Yen T-T, Chen I-C, Hua M-W, Wei C-Y, Shih K-H, Li J-L, Lin C-H, Hsiao T-H, Chen Y-M, Jiang R-S. A KCNQ4 c.546C>G Genetic Variant Associated with Late Onset Non-Syndromic Hearing Loss in a Taiwanese Population. Genes. 2021; 12(11):1711. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111711
Chicago/Turabian StyleYen, Ting-Ting, I-Chieh Chen, Men-Wei Hua, Chia-Yi Wei, Kai-Hsiang Shih, Jui-Lin Li, Ching-Heng Lin, Tzu-Hung Hsiao, Yi-Ming Chen, and Rong-San Jiang. 2021. "A KCNQ4 c.546C>G Genetic Variant Associated with Late Onset Non-Syndromic Hearing Loss in a Taiwanese Population" Genes 12, no. 11: 1711. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111711