
Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
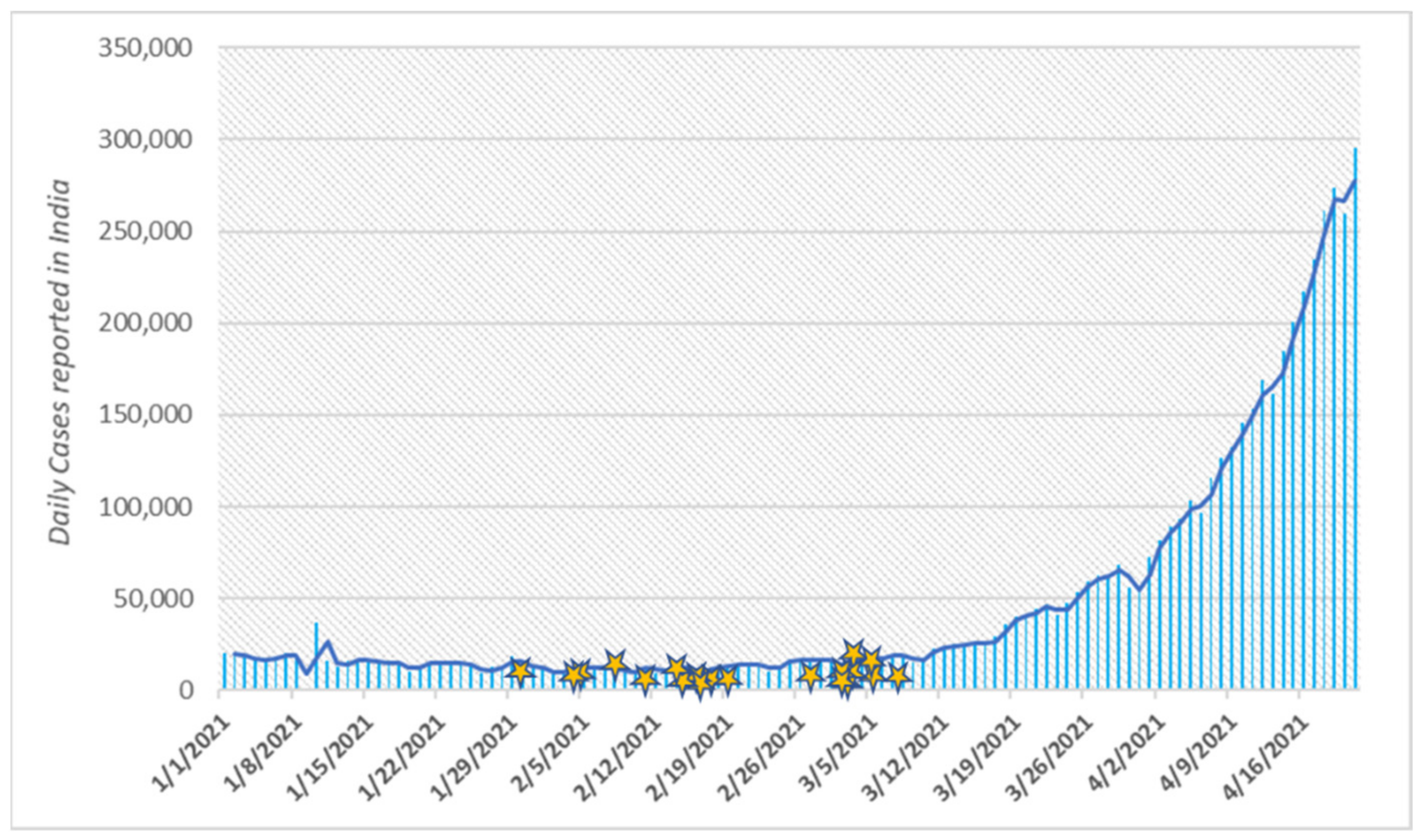
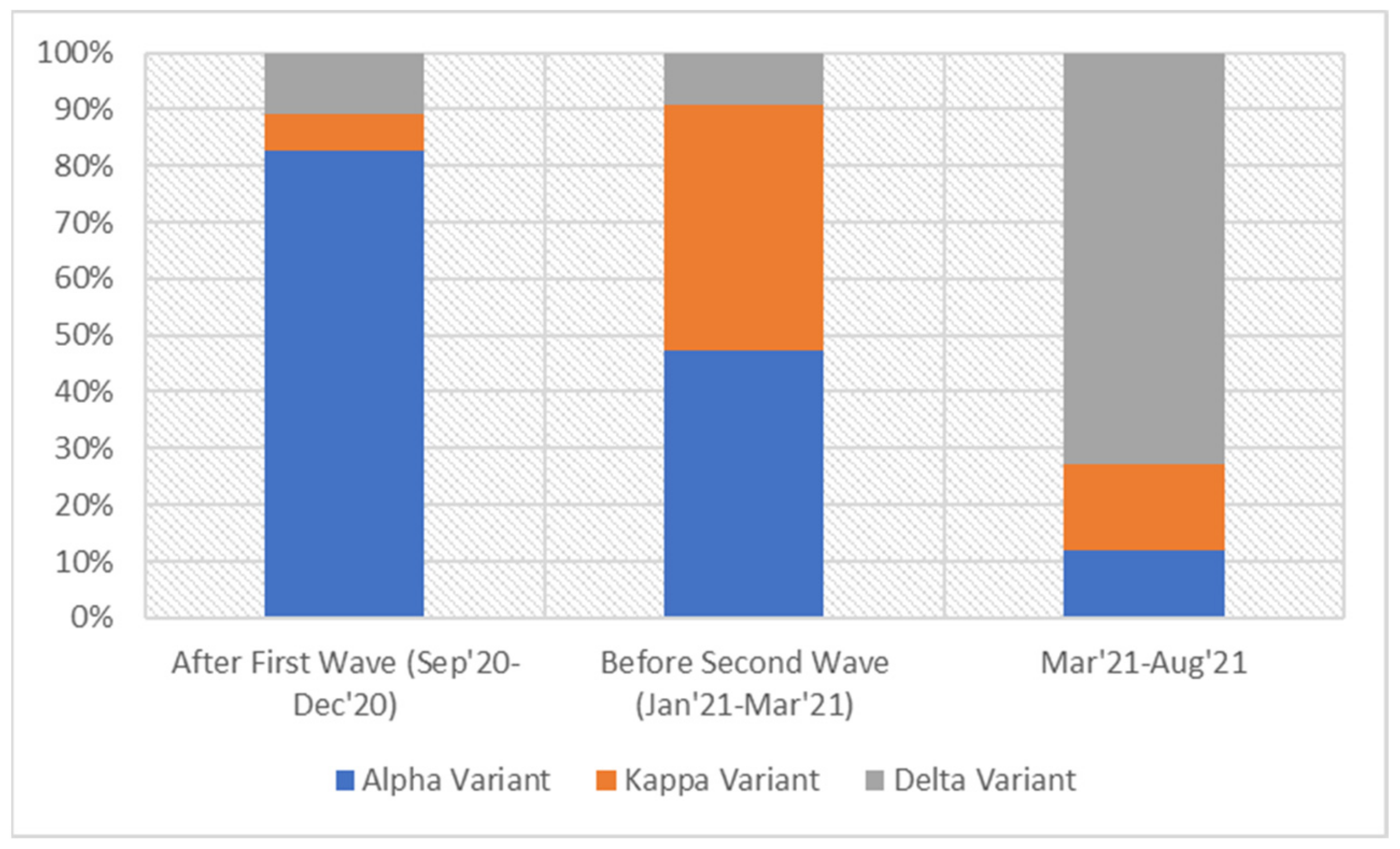
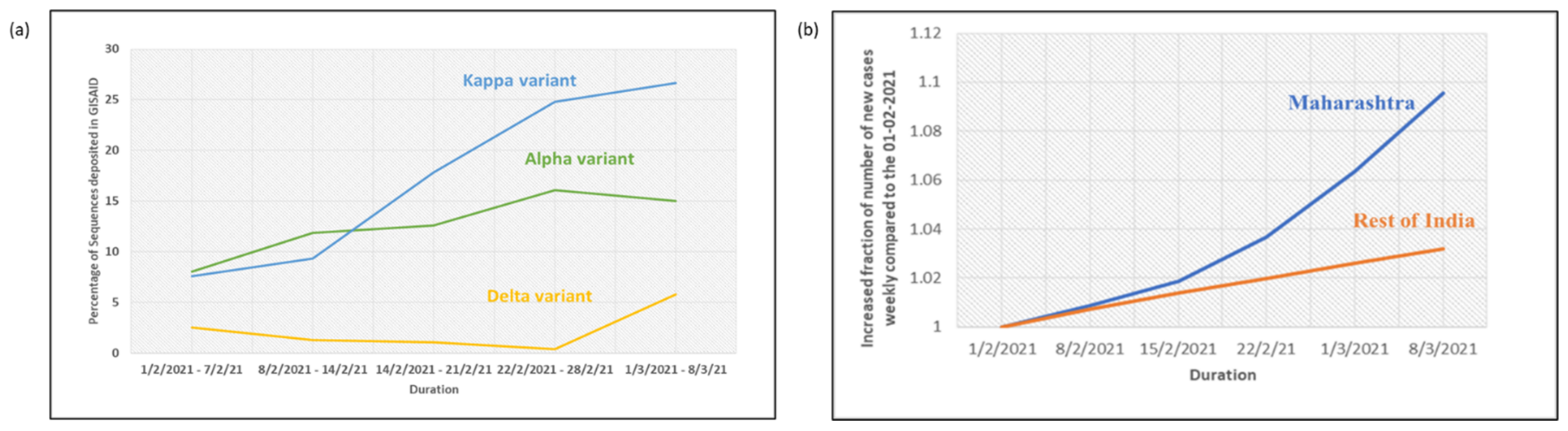
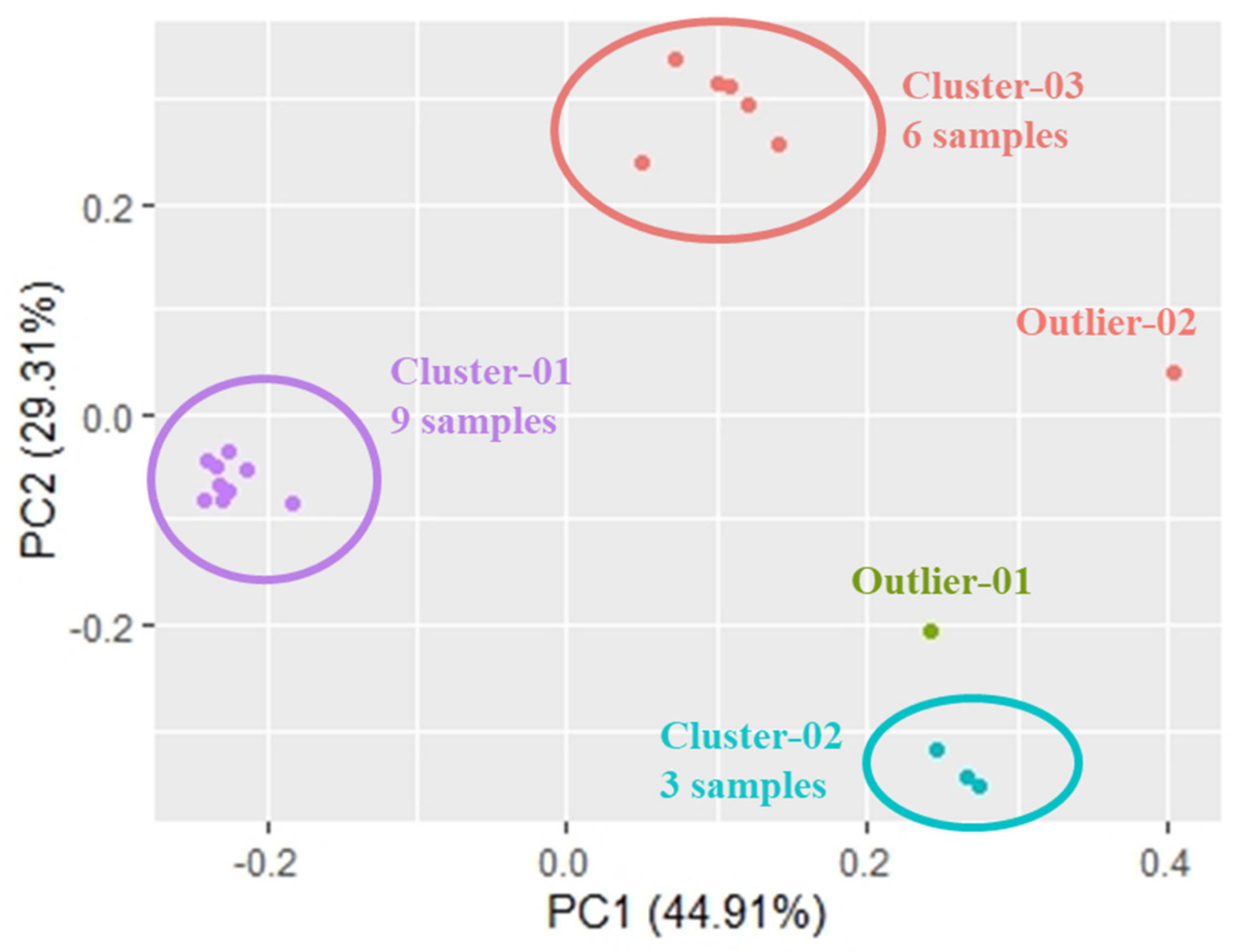
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO COVID-19 Dashboard. 2020. Available online: https://covid19.who.int/ (accessed on 26 April 2021).
- World Health Organization. Novel Coronavirus (2019-nCoV), Weekly Epidemiological Update on COVID-19. 20 April 2021. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20210420_weekly_epi_update_36.pdf?sfvrsn=ab75add5_7&download=true (accessed on 30 January 2020).
- Asrani, P.; Eapen, M.S.; Hassan, M.I.; Sohal, S.S. Implications of the second wave of COVID-19 in India. Lancet Respir. Med. 2021, 9, e93–e94. [Google Scholar] [CrossRef]
- De Souza, F.S.H.; Hojo-Souza, N.S.; da Silva, C.M.; Guidoni, D.L. Second wave of COVID-19 in Brazil: Younger at higher risk. Eur. J. Epidemiol. 2021, 36, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Iftimie, S.; López-Azcona, A.F.; Vallverdú, I.; Hernández-Flix, S.; de Febrer, G.; Parra, S.; Hernández-Aguilera, A.; Riu, F.; Joven, J.; Andreychuk, N.; et al. First and second waves of coronavirus disease-19: A comparative study in hospitalized patients in Reus, Spain. PLoS ONE 2021, 16, e0248029. [Google Scholar] [CrossRef] [PubMed]
- Salyer, S.J.; Maeda, J.; Sembuche, S.; Kebede, Y.; Tshangela, A.; Moussif, M.; Ihekweazu, C.; Mayet, N.; Abate, E.; Ouma, A.O.; et al. The first and second waves of the COVID-19 pandemic in Africa: A cross-sectional study. Lancet 2021, 397, 1265–1275. [Google Scholar] [CrossRef]
- Ion PITM Hi-QTM Sequencing 200 Kit. Available online: http://tools.thermofisher.com/content/sfs/manuals/MAN0010947_Ion_PI_HiQ_Seq_200_Kit_UG.pdf (accessed on 6 July 2021).
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team R core team. R: A Language and Environment for Statistical Computing 2021. Available online: http//www.R-project.org (accessed on 1 August 2021).
- GISAID. GISAID Initiative. Adv. Virus Res. 2020, 2008, 1–7. [Google Scholar]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Toovey, O.T.R.; Harvey, K.N.; Bird, P.W.; Tang, J.W.-T.W.-T. Introduction of Brazilian SARS-CoV-2 484K.V2 related variants into the UK. J. Infect. 2021, 82, e23–e24. [Google Scholar] [CrossRef] [PubMed]
- Annavajhala, M.K.; Mohri, H.; Zucker, J.E.; Sheng, Z.; Wang, P.; Gomez-Simmonds, A.; Ho, D.D.; Uhlemann, A.-C. A Novel SARS-CoV-2 Variant of Concern, B.1.526, Identified in New York. medRxiv Prepr. Serv. Health Sci. 2021. [Google Scholar] [CrossRef]
- Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. Available online: https://www.fda.gov/media/145802/download (accessed on 24 August 2021).
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. 2020, 1–9. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 27 September 2021).
- Zhang, W.; Davis, B.D.; Chen, S.S.; Sincuir Martinez, J.M.; Plummer, J.T.; Vail, E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA 2021, 325, 1324. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.Z.; Paul, S.K.; Begum, J.A.; Nasreen, S.A.; Ahmed, S.; Ahmad, F.U.; Aziz, M.A.; Parvin, R.; Aung, M.S.; Kobayashi, N. Extensive genetic diversity with novel mutations in spike glycoprotein of severe acute respiratory syndrome coronavirus 2, Bangladesh in late 2020. New Microbes. New Infect. 2021, 41, 100889. [Google Scholar] [CrossRef] [PubMed]
- Khateeb, J.; Li, Y.; Zhang, H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care 2021, 25, 244. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R. Present variants of concern and variants of interest of severe acute respiratory syndrome coronavirus 2: Their significant mutations in S-glycoprotein, infectivity, re-infectivity, immune escape and vaccines activity. Rev. Med. Virol. 2021, e2270. [Google Scholar] [CrossRef]
- Chen, L.-L.; Lu, L.; Choi, C.Y.-K.; Cai, J.-P.; Tsoi, H.-W.; Chu, A.W.-H.; Ip, J.D.; Chan, W.-M.; Zhang, R.R.; Zhang, X.; et al. Impact of SARS-CoV-2 variant-associated RBD mutations on the susceptibility to serum antibodies elicited by COVID-19 infection or vaccination. Clin. Infect. Dis. 2021, ciab656. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.-W. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun. Biol. 2021, 4, 228. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Addressable Wells | Chip-1 (9 Samples) | Chip-2 (11 Samples) | ||||
|---|---|---|---|---|---|---|
| 37,849,615 | 37,849,615 | |||||
| With ISPs | 33,700,206 | 89.00% | 32,798,330 | 86.70% | ||
| Live | 33,698,044 | 100.00% | 32,749,088 | 99.80% | ||
| Test Fragments | 680,001 | 2.00% | 657,319 | 2.00% | ||
| Library | 33,018,043 | 98.00% | 32,091,769 | 98.00% | ||
| Library ISPs | 33,018,043 | 32,091,769 | ||||
| Filtered: Polyclonal | 8,159,862 | 24.70% | 7,284,699 | 22.70% | ||
| Filtered: Low Quality | 22,21,791 | 6.70% | 1,630,141 | 5.10% | ||
| Filtered: Adapter Dimer | 56,745 | 0.20% | 158,413 | 0.50% | ||
| Final Library ISPs | 22,579,645 | 68.40% | 23,018,516 | 71.70% | ||
| Total Reads | 22,391,814 | 22,825,334 | ||||
| Aligned Reads | 21,175,935 | 94.60% | 22,067,308 | 96.70% | ||
| Unaligned Reads | 1,215,879 | 5.40% | 758,026 | 3.30% | ||
| Alignment Quality | AQ17 | AQ20 | Perfect | AQ17 | AQ20 | Perfect |
| Total Number of Bases [Mbp] | 3.83 G | 3.68 G | 2.96 G | 4.18 G | 4.02 G | 3.25 G |
| Mean Length [bp] | 193 | 189 | 158 | 197 | 192 | 160 |
| Longest Alignment [bp] | 377 | 376 | 359 | 343 | 343 | 332 |
| Mean Coverage Depth | 57.1 | 54.8 | 44.1 | 62.3 | 59.8 | 48.4 |
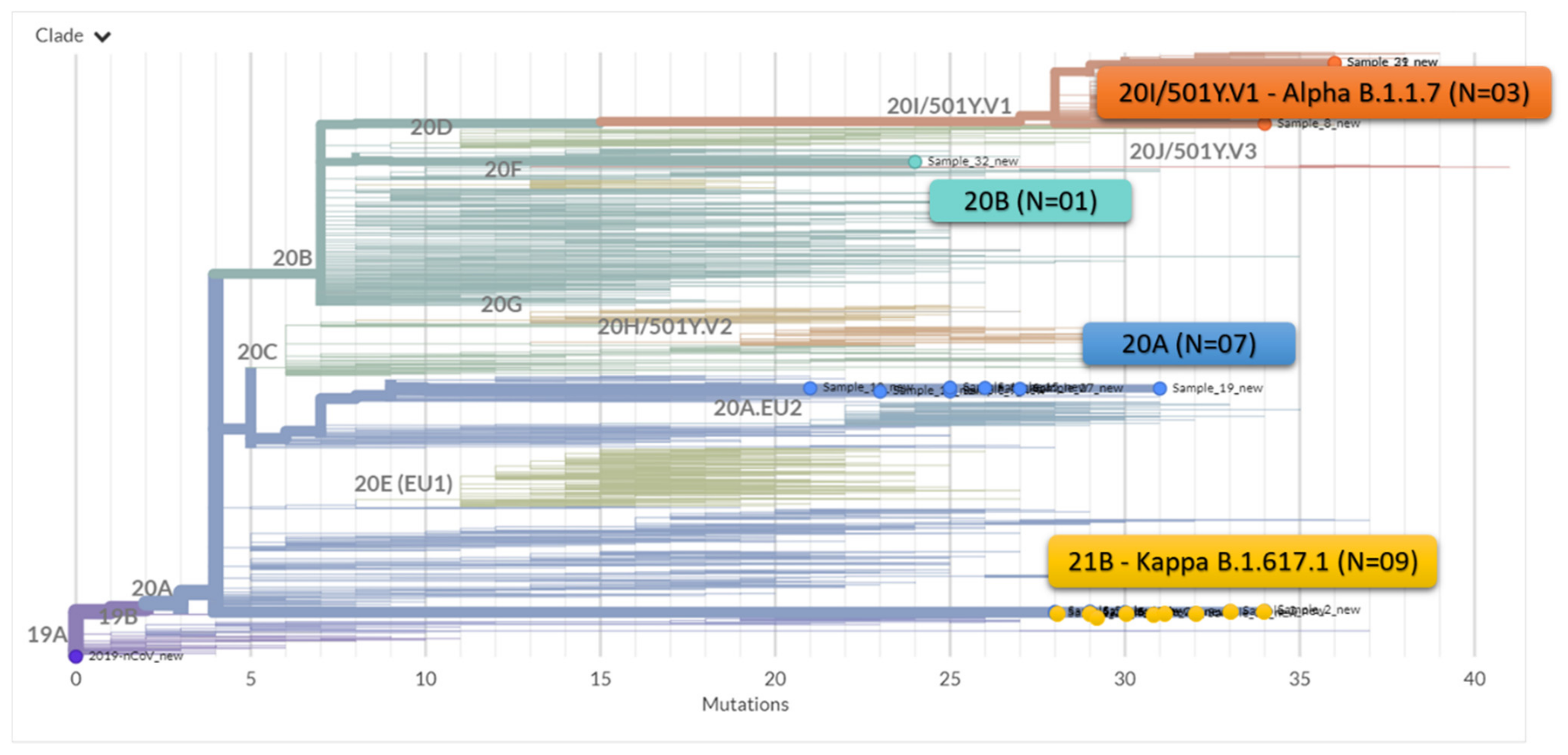
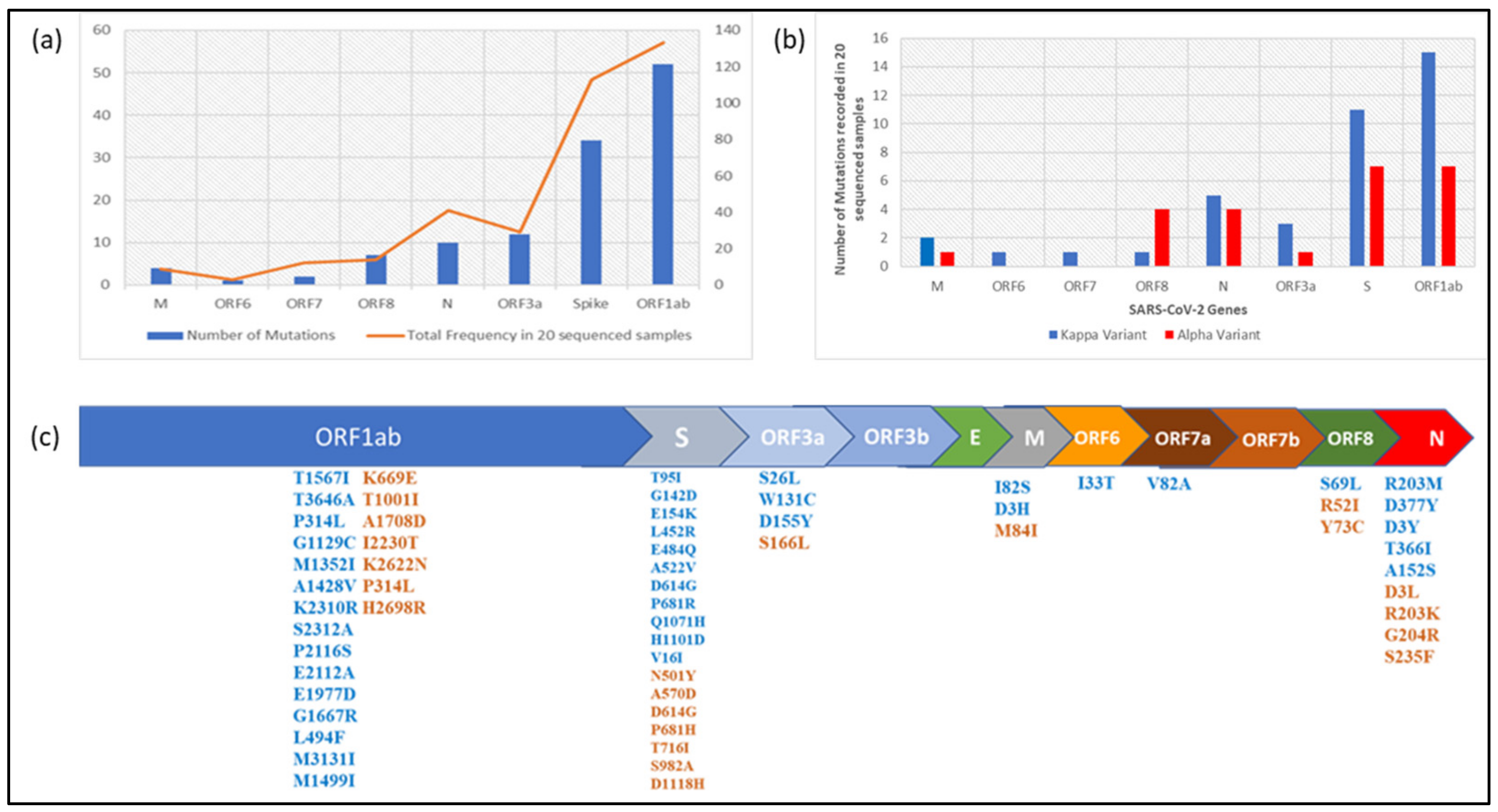
| S.No. | Patient ID | Qbit 4.O Reading ng/ul-RNA Sample | GenBank Accession No. | GISAID Accession No. | No. of Mapped Reads | Total Assigned Amplicon Reads | % on Target Reads | Average Read per Amplicon | Mean Depth | %Uniformity | Assembly Length (bp) | PANGO Lineage | Emerging Clade | GISAID Clade |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NCOV/AB/21/FL407 | 3.02 | MZ562746 | EPI_ISL_1972141 | 1,378,010 | 1,364,626 | 99.03 | 5758 | 7503 | 74.30% | 29,837 | B.1.617.1 | 21B (Kappa) | G |
| 2 | NCOV/AB/21/FL409 | 8.87 | MZ562747 | EPI_ISL_1972134 | 3,733,707 | 3,732,055 | 99.96 | 15,747 | 25,230 | 97.89% | 29,833 | B.1.617.1 | 21B (Kappa) | G |
| 3 | NCOV/AB/21/FL901 | 5.92 | MZ562748 | EPI_ISL_1972135 | 2,389,121 | 2,388,613 | 99.98 | 10,079 | 15,986 | 97.03% | 29,834 | B.1.617.1 | 21B (Kappa) | G |
| 4 | NCOV/AB/21/FL902 | 5.53 | MZ562749 | EPI_ISL_1972136 | 2,725,685 | 2,694,642 | 98.86 | 11,370 | 18,478 | 99.29% | 29,840 | B.1.617.1 | 21B (Kappa) | G |
| 5 | NCOV/AB/21/FL832 | 6.73 | MZ562750 | EPI_ISL_1972130 | 661,367 | 144,412 | 21.84 | 609.3 | 936.4 | 99.28% | 29,816 | B.1.1.7 | 20I/501Y.V1 | GRY |
| 6 | NCOV/AB/21/FL405 | 3.72 | MZ562751 | EPI_ISL_1972142. | 3,581,768 | 3,544,684 | 98.96 | 14,956 | 22,246 | 97.10% | 29,826 | B.1.36 | 20A | GH |
| 7 | NCOV/AB/21/EX986 | 3.24 | MZ723921 | EPI_ISL_3305853 | 394,294 | 371,849 | 94.31 | 1569 | 2,473 | 97.28% | 29,800 | B.1.36 | 20A | GH |
| 8 | NCOV/AB/20/CA052 | 5.65 | MZ562752 | EPI_ISL_1972143 | 4,349,146 | 4,289,977 | 98.64 | 18,101 | 28,961 | 99.54% | 29,834 | B.1.36 | 20A | GH |
| 9 | NCOV/AB/21/FA012 | 6.69 | MZ562753 | EPI_ISL_1972144 | 1,506,394 | 1,446,477 | 96.02 | 6103 | 9750 | 99.49% | 29,848 | B.1.36 | 20A | GH |
| 10 | NCOV/AB/21/FB757 | 3.73 | MZ562754 | EPI_ISL_1972145 | 4,124,452 | 4,104,495 | 99.52 | 17,319 | 26,603 | 96.98% | 29,834 | B.1.36 | 20A | GH |
| 11 | NCOV/AB/21/FD308 | 3.88 | MZ562755 | EPI_ISL_1972146 | 80,774 | 79,290 | 98.16 | 334.6 | 534.7 | 99.12% | 29,834 | B.1.36 | 20A | GH |
| 12 | NCOV/AB/21/FF663 | 3.38 | MZ562756 | EPI_ISL_1972137 | 393,314 | 275,581 | 70.07 | 1163 | 1,842 | 98.12% | 29,833 | B.1.617.1 | 21B (Kappa) | G |
| 13 | NCOV/AB/21/FF945 | 4.34 | MZ562757 | EPI_ISL_1972138 | 2,080,768 | 2,058,700 | 98.94 | 8686 | 13,990 | 98.64% | 29,836 | B.1.617.1 | 21B (Kappa) | G |
| 14 | NCOV/AB/21/EQ324 | 5.32 | MZ562758 | EPI_ISL_1972139 | 917,154 | 899,993 | 98.13 | 3797 | 5,763 | 97.97% | 29,832 | B.1.617.1 | 21B (Kappa) | G |
| 15 | NCOV/AB/21/FG788 | 3.34 | MZ562759 | EPI_ISL_1972140 | 3,939,432 | 3,917,148 | 99.43 | 16,528 | 26,628 | 98.21% | 29,835 | B.1.617.1 | 21B (Kappa) | G |
| 16 | NCOV/AB/21/FK354 | 7.18 | MZ562760 | EPI_ISL_1972147 | 1,945,720 | 1,901,249 | 97.71 | 8022 | 12,758 | 99.11% | 29,853 | B.1.36 | 20A | GH |
| 17 | NCOV/AB/21/FL862 | 4.35 | MZ723922 | EPI_ISL_1972132 | 3,313,340 | 3,203,191 | 96.68 | 13,516 | 21,669 | 97.89% | 29,823 | B.1.1.7 | 20I/501Y.V1 | GRY |
| 18 | NCOV/AB/21/FK940 | 4.94 | MZ562761 | EPI_ISL_1972133 | 2,262,299 | 2,261,261 | 99.95 | 9541 | 14,961 | 99.10% | 29,840 | B.1.617.1 | 21B (Kappa) | G |
| 19 | NCOV/AB/21/FL880 | 5.27 | MZ562762 | EPI_ISL_1972131 | 2,668,661 | 2,667,095 | 99.94 | 11,254 | 18,319 | 99.06% | 29,817 | B.1.1.7 | 20I/501Y.V1 | GRY |
| 20 | NCOV/AB/21/FL414 | 6.54 | MZ734487 | EPI_ISL_3316398 | 238,608 | 125,526 | 52.61 | 529.6 | 848 | 92.73% | 29,834 | B.1.306 | 20B | GR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, J.; Malhotra, A.G.; Biswas, D.; Shankar, P.; Lokhande, L.; Yadav, A.K.; Raghuvanshi, A.; Kale, D.; Nema, S.; Saigal, S.; et al. Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India. Genes 2021, 12, 1803. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111803
Singh J, Malhotra AG, Biswas D, Shankar P, Lokhande L, Yadav AK, Raghuvanshi A, Kale D, Nema S, Saigal S, et al. Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India. Genes. 2021; 12(11):1803. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111803
Chicago/Turabian StyleSingh, Jitendra, Anvita Gupta Malhotra, Debasis Biswas, Prem Shankar, Leena Lokhande, Ashvini Kumar Yadav, Arun Raghuvanshi, Dipesh Kale, Shashwati Nema, Saurabh Saigal, and et al. 2021. "Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India" Genes 12, no. 11: 1803. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111803