
Genetic Diversity and Population Structure Analysis of the USDA Olive Germplasm Using Genotyping-By-Sequencing (GBS)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and Genotyping-By-Sequencing (GBS) Procedures
2.3. Data Analysis
2.3.1. SNP Discovery
2.3.2. SNP Marker Properties
2.3.3. Population Molecular Characterization
3. Results
3.1. Characterization and Distribution of the GBS-Generated SNPs in the Olive Genomes
3.2. Characterization of the SNP Markers
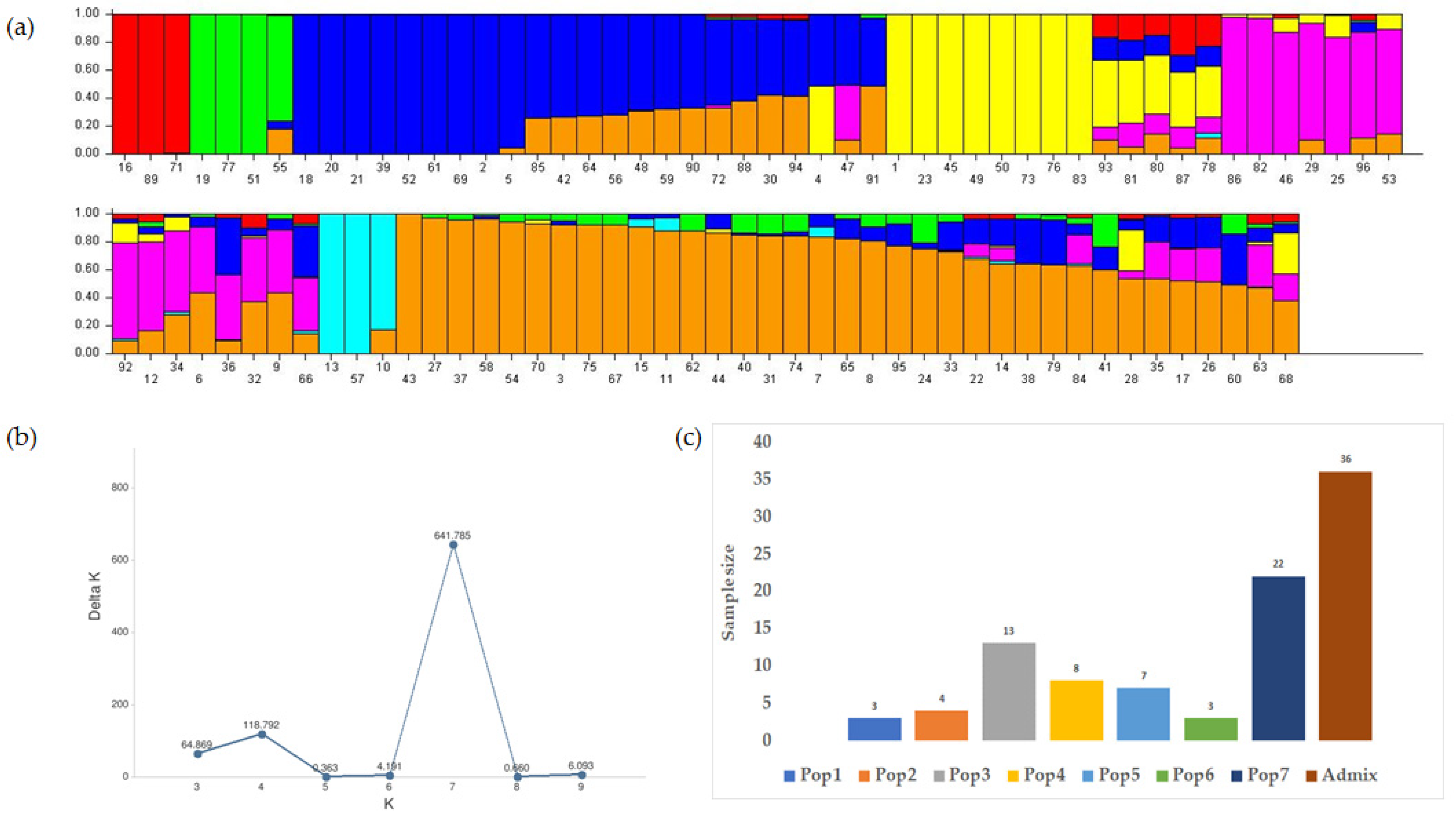
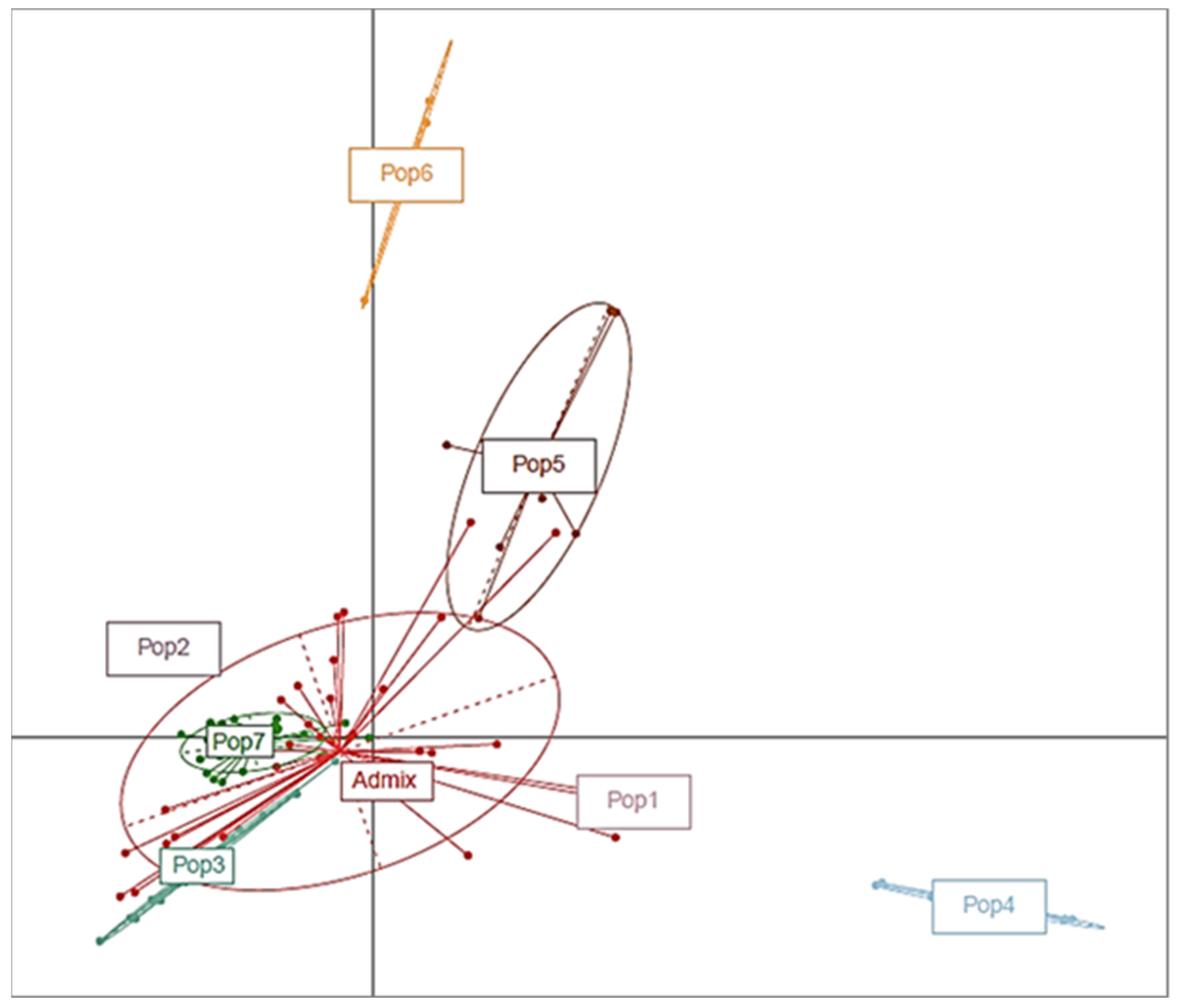
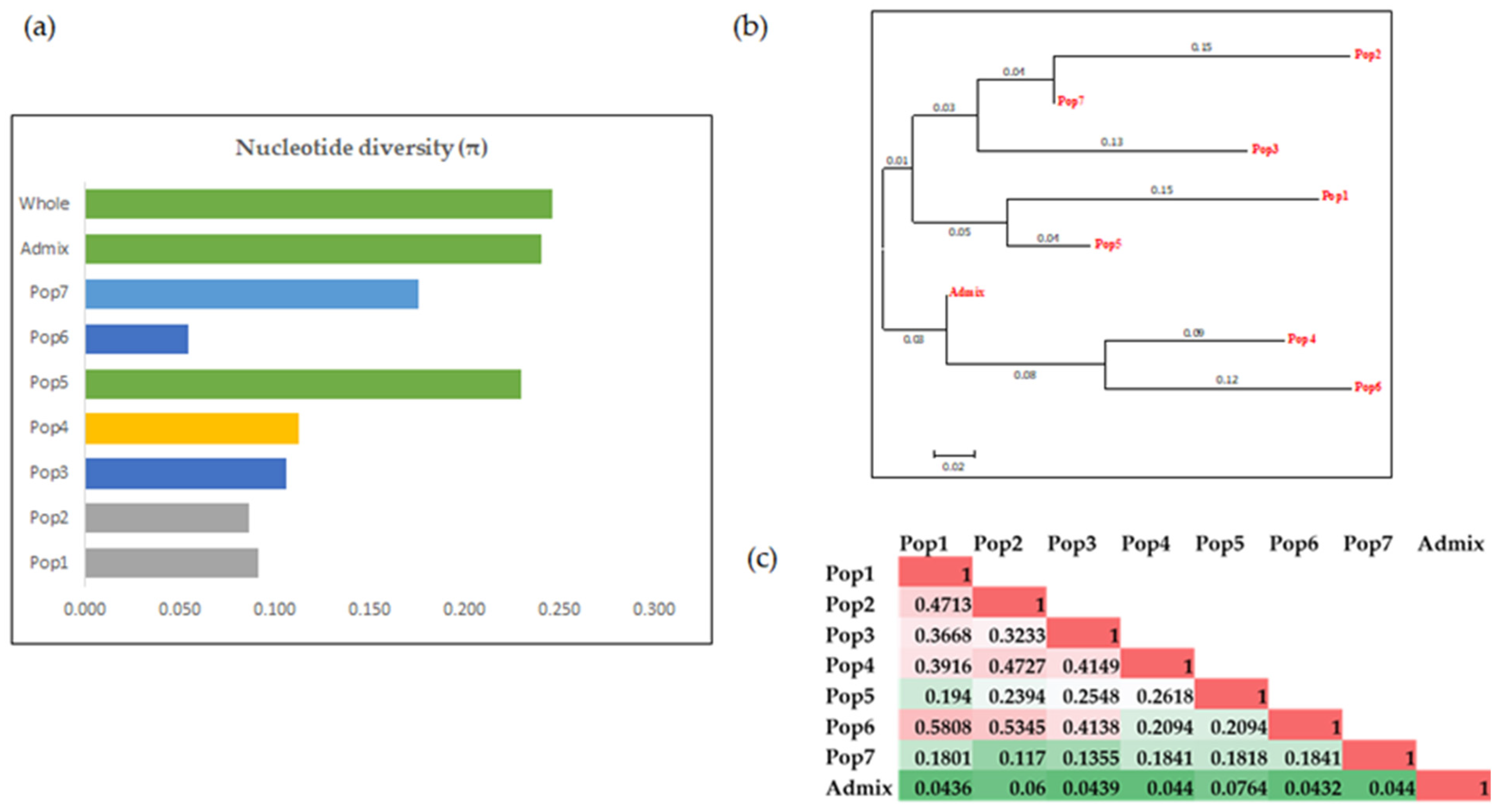
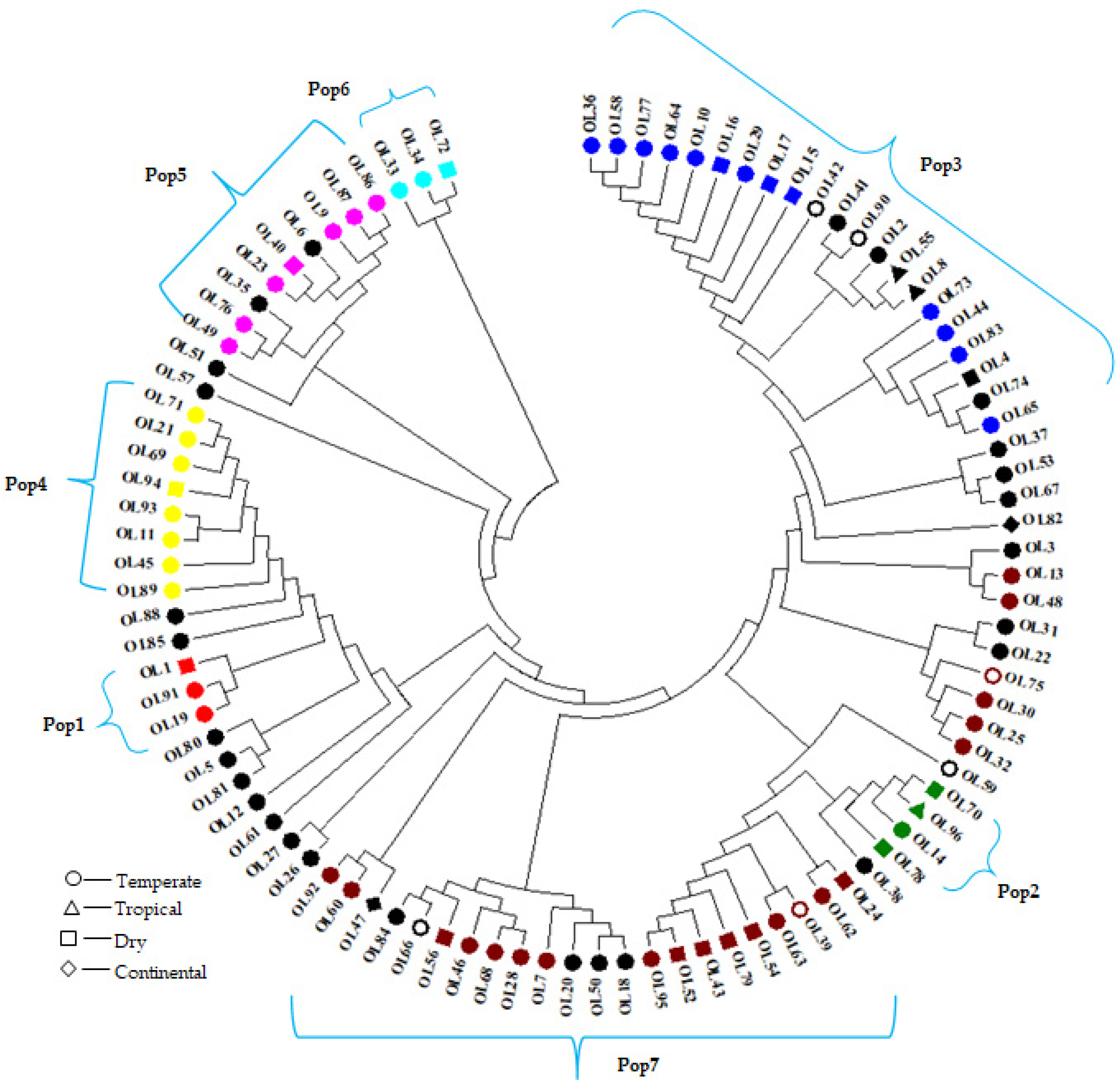
3.3. Characterization of the Population and the Genetic Relationships
3.3.1. Structure Analysis and the Genetic Relationships
3.3.2. Cluster Analysis
3.3.3. Analysis of Molecular Variance (AMOVA)
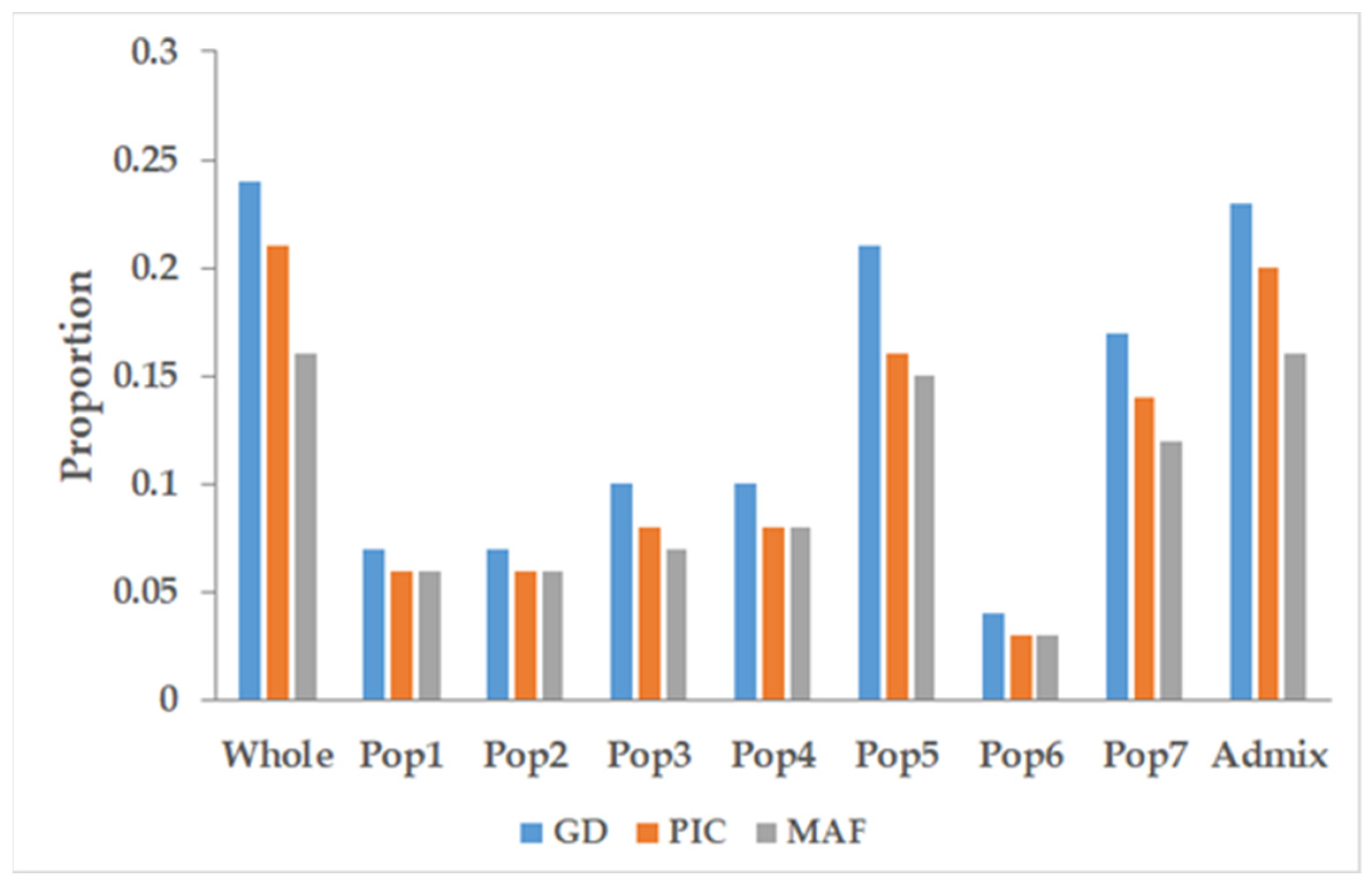
3.4. Marker Characteristics across the Populations
4. Discussion
4.1. GBS Analysis of the Olive Genomes
4.2. Features of the SNP Markers
4.3. Features of the US Repository Olive Population
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sebastiani, L.; Busconi, M. Recent developments in olive (Olea europaea L.) genetics and genomics: Applications in taxonomy, varietal identification, traceability and breeding. Plant Cell Rep. 2017, 36, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Gouvinhas, I.; Machado, N.; Sobreira, C.; Domínguez-Perles, R.; Gomes, S.; Rosa, E.; Barros, A.I.R.N.A. Critical Review on the Significance of Olive Phytochemicals in Plant Physiology and Human Health. Molecules 2017, 22, 1986. [Google Scholar] [CrossRef] [Green Version]
- Kaya, H.B.; Akdemir, D.; Lozano, R.; Cetin, O.; Kaya, H.S.; Sahin, M.; Smith, J.L.; Tanyolac, B.; Jannink, J.-L. Genome wide association study of 5 agronomic traits in olive (Olea europaea L.). Sci. Rep. 2019, 9, 18764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Miranda, J.; Pérez-Jiménez, F.; Ros, E.; De Caterina, R.; Badimón, L.; Covas, M.; Escrich, E.; Ordovás, J.; Soriguer, F.; Abiá, R.; et al. Olive oil and health: Summary of the II international conference on olive oil and health consensus report, Jaén and Córdoba (Spain) 2008. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Niu, E.; Shi, A.; Mou, B. Genetic Diversity Analysis of Olive Germplasm (Olea europaea L.) With Genotyping-by-Sequencing Technology. Front. Genet. 2019, 10, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniewski, D.; Van Campo, E.; Boiy, T.; Terral, J.-F.; Khadari, B.; Besnard, G. Primary domestication and early uses of the emblematic olive tree: Palaeobotanical, historical and molecular evidence from the Middle East. Biol. Rev. 2012, 87, 885–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Agricultural Statistics Service (NASS); U.S.A. Department of Agriculture. Noncitrus Fruits and Nuts Summary. Available online: https://downloads.usda.library.cornell.edu/usda-esmis/files/zs25x846c/sf269213r/6t054c23t/ncit0521.pdf (accessed on 11 November 2021).
- Lanner, R.M.; Taylor, J.M. The Olive in California: History of an Immigrant Tree. West. Hist. Q. 2002, 33, 494. [Google Scholar] [CrossRef]
- Lavanya, G.R.; Srivastava, J.; Ranade, S.A. Molecular assessment of genetic diversity in mung bean germplasm. J. Genet. 2008, 87, 65–74. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, N.; Taranto, F.; Camposeo, S.; Mangini, G.; Fanelli, V.; Gadaleta, S.; Miazzi, M.M.; Pavan, S.; Di Rienzo, V.; Sabetta, W.; et al. GBS-derived SNP catalogue unveiled wide genetic variability and geographical relationships of Italian olive cultivars. Sci. Rep. 2018, 8, 15877. [Google Scholar] [CrossRef] [Green Version]
- Ipek, A.; Yılmaz, K.; Sıkıcı, P.; Tangu, N.A.; Öz, A.T.; Bayraktar, M.; Ipek, M.; Gülen, H. SNP Discovery by GBS in Olive and the Construction of a High-Density Genetic Linkage Map. Biochem. Genet. 2016, 54, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Sion, S.; Savoia, M.A.; Gadaleta, S.; Piarulli, L.; Mascio, I.; Fanelli, V.; Montemurro, C.; Miazzi, M.M. How to Choose a Good Marker to Analyze the Olive Germplasm (Olea europaea L.) and Derived Products. Genes 2021, 12, 1474. [Google Scholar] [CrossRef]
- Pontikis, C.A.; Loukas, M.; Kousounis, G. The use of Biochemical Markers to Distinguish Olive Cultivars. J. Hortic. Sci. 1980, 55, 333–343. [Google Scholar] [CrossRef]
- Decroocq, S.; Cornille, A.; Tricon, D.; Babayeva, S.; Chague, A.; Eyquard, J.-P.; Karychev, R.; Dolgikh, S.; Kostritsyna, T.; Liu, S.; et al. Data from: New insights into the history of domesticated and wild apricots and its contribution to Plum pox virus resistance. Mol. Ecol. 2016, 25, 4712–4729. [Google Scholar] [CrossRef] [PubMed]
- Delplancke, M.; Alvarez, N.; Benoit, L.; Espindola, A.; Joly, H.; Neuenschwander, S.; Arrigo, N. Evolutionary history of almond tree domestication in the M editerranean basin. Mol. Ecol. 2013, 22, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.M.; Volk, G.M.; Reilley, A.A.; Henk, A.D.; Lockwood, D.R.; Reeves, P.A.; Forsline, P.L. Genetic diversity and population structure in Malus sieversii, a wild progenitor species of domesticated apple. Tree Genet. Genomes 2009, 5, 339–347. [Google Scholar] [CrossRef]
- Kumar, S.; Garrick, D.J.; Bink, M.C.; Whitworth, C.; Chagné, D.; Volz, R.K. Novel genomic approaches unravel genetic architecture of complex traits in apple. BMC Genom. 2013, 14, 393. [Google Scholar] [CrossRef] [Green Version]
- Kouassi, A.B.; Durel, C.-E.; Costa, F.; Tartarini, S.; van de Weg, E.; Evans, K.; Fernandez-Fernandez, F.; Govan, C.; Boudichevskaja, A.; Dunemann, F.; et al. Estimation of genetic parameters and prediction of breeding values for apple fruit-quality traits using pedigreed plant material in Europe. Tree Genet. Genomes 2009, 5, 659–672. [Google Scholar] [CrossRef]
- Luo, Z.; Brock, J.; Dyer, J.M.; Kutchan, T.; Schachtman, D.; Augustin, M.; Ge, Y.; Fahlgren, N.; Abdel-Haleem, H. Genetic Diversity and Population Structure of a Camelina sativa Spring Panel. Front. Plant Sci. 2019, 10, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.M.; Winfield, M.O.; Burridge, A.J.; Downie, R.C.; Benbow, H.R.; Barker, G.L.A.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; et al. Characterization of a Wheat Breeders’ Array suitable for high-throughput SNP genotyping of global accessions of hexaploid bread wheat (Triticum aestivum). Plant Biotechnol. J. 2017, 15, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Yuan, Y.; Wang, H.; Yu, D.; Liu, Y.; Zhang, A.; Gowda, M.; Nair, S.K.; Hao, Z.; Lu, Y.; et al. Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci. Rep. 2020, 10, 16308. [Google Scholar] [CrossRef] [PubMed]
- Kitony, J.; Sunohara, H.; Tasaki, M.; Mori, J.-I.; Shimazu, A.; Reyes, V.; Yasui, H.; Yamagata, Y.; Yoshimura, A.; Yamasaki, M.; et al. Development of an Aus-Derived Nested Association Mapping (Aus-NAM) Population in Rice. Plants 2021, 10, 1255. [Google Scholar] [CrossRef]
- McNally, K.L.; Childs, K.L.; Bohnert, R.; Davidson, R.M.; Zhao, K.; Ulat, V.J.; Zeller, G.; Clark, R.M.; Hoen, D.R.; Bureau, T.E.; et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. USA 2009, 106, 12273–12278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, H.; Shimada, T.; Nonaka, K.; Kita, M.; Kuniga, T.; Endo, T.; Ikoma, Y.; Omura, M. High-throughput genotyping in citrus accessions using an SNP genotyping array. Tree Genet. Genomes 2013, 9, 145–153. [Google Scholar] [CrossRef]
- Micheletti, D.; Dettori, M.T.; Micali, S.; Aramini, V.; Pacheco, I.; Linge, C.D.S.; Foschi, S.; Banchi, E.; Barreneche, T.; Quilot-Turion, B.; et al. Whole-Genome Analysis of Diversity and SNP-Major Gene Association in Peach Germplasm. PLoS ONE 2015, 10, e0136803. [Google Scholar] [CrossRef]
- Larsen, B.; Gardner, K.; Pedersen, C.; Ørgaard, M.; Migicovsky, Z.; Myles, S.; Toldam-Andersen, T. Population structure, relatedness and ploidy levels in an apple gene bank revealed through genotyping-by-sequencing. PLoS ONE 2018, 13, e0201889. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Luo, T.; Zhang, W.; Mason, A.; Huang, D.; Huang, X.; Tang, W.; Dou, Y.; Zhang, C.; Xiao, Y. Development of High-Density SNP Markers and Their Application in Evaluating Genetic Diversity and Population Structure in Elaeis guineensis. Front. Plant Sci. 2019, 10, 130. [Google Scholar] [CrossRef]
- Koehmstedt, A.M.; Aradhya, M.K.; Soleri, D.; Smith, J.L.; Polito, V.S. Molecular characterization of genetic diversity, structure, and differentiation in the olive (Olea europaea L.) germplasm collection of the United States Department of Agriculture. Genet. Resour. Crop. Evol. 2010, 58, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Kamvar, Z.N.; Collins, C.; Lustrik, R.; Beugin, M.-P.; Knaus, B.J.; Jombart, M.T. Package ‘Adegenet’. Github Repository. Available online: https://github.com/thibautjombart/adegenet (accessed on 11 November 2021).
- Kottek, M.; Grieser, J.; Beck, C.; Rudolf, B.; Rubel, F. World Map of the Köppen-Geiger climate classification updated. Meteorol. Z. 2006, 15, 259–263. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Julca, I.; Marcet-Houben, M.; Cruz, F.; Gómez-Garrido, J.; Gaut, B.S.; Díez, C.M.; Gut, I.G.; Alioto, T.S.; Vargas, P.; Gabaldón, T. Genomic evidence for recurrent genetic admixture during the domestication of Mediterranean olive trees (Olea europaea L.). BMC Biol. 2020, 18, 148. [Google Scholar] [CrossRef] [PubMed]
- Nardi, E.P.; Evangelista, F.S.; Tormen, L.; Saint’pierre, T.D.; Curtius, A.J.; de Souza, S.S.; Barbosa, F. The use of inductively coupled plasma mass spectrometry (ICP-MS) for the determination of toxic and essential elements in different types of food samples. Food Chem. 2009, 112, 727–732. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Granato, I.S.C.; Galli, G.; Couto, E.G.O.; Souza, M.B.; Mendonça, L.F.; Fritsche-Neto, R. snpReady: A tool to assist breeders in genomic analysis. Mol. Breed. 2018, 38, 102. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. In Genetics; 2000; 155, pp. 945–959. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/10835412 (accessed on 11 November 2021). [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Belaj, A.; Satovic, Z.; Rallo, L.; Trujillo, I. Genetic diversity and relationships in olive (Olea europaea L.) germplasm collections as determined by randomly amplified polymorphic DNA. Theor. Appl. Genet. 2002, 105, 638–644. [Google Scholar] [CrossRef]
- Besnard, G.; Baradat, P.; Bervillé, A. Genetic relationships in the olive (Olea europaea L.) reflect multilocal selection of cultivars. Theor. Appl. Genet. 2001, 102, 251–258. [Google Scholar] [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.K.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [Green Version]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-sequencing approaches to characterize crop genomes: Choosing the right tool for the right application. Plant Biotechnol. J. 2017, 15, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, F.; Julca, I.; Gómez-Garrido, J.; Loska, D.; Marcet-Houben, M.; Cano, E.; Galán, B.; Frias, L.; Ribeca, P.; Derdak, S.; et al. Genome sequence of the olive tree, Olea europaea. GigaScience 2016, 5, 29. [Google Scholar] [CrossRef]
- Mantello, C.C.; Cardoso-Silva, C.B.; Da Silva, C.C.; De Souza, L.M.; Junior, E.J.S.; Gonçalves, P.D.S.; Vicentini, R.; De Souza, A.P. De Novo Assembly and Transcriptome Analysis of the Rubber Tree (Hevea brasiliensis) and SNP Markers Development for Rubber Biosynthesis Pathways. PLoS ONE 2014, 9, e102665. [Google Scholar] [CrossRef] [Green Version]
- Clarke, W.E.; Parkin, I.A.; Gajardo, H.A.; Gerhardt, D.J.; Higgins, E.; Sidebottom, C.; Sharpe, A.G.; Snowdon, R.J.; Federico, M.L.; Iniguez-Luy, F.L. Genomic DNA enrichment using sequence capture microarrays: A novel approach to discover sequence nucleotide polymorphisms (SNP) in Brassica napus L. PLoS ONE 2013, 8, e81992. [Google Scholar]
- Morton, B.R.; Bi, I.V.; McMullen, M.D.; Gaut, B.S. Variation in Mutation Dynamics Across the Maize Genome as a Function of Regional and Flanking Base Composition. Genetics 2006, 172, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shete, S.; Tiwari, H.; Elston, R.C. On Estimating the Heterozygosity and Polymorphism Information Content Value. Theor. Popul. Biol. 2000, 57, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Belaj, A.; De La Rosa, R.; Lorite, I.J.; Mariotti, R.; Cultrera, N.G.; Beuzón, C.R.; González-Plaza, J.J.; Muñoz-Merida, A.; Trelles, O.; Baldoni, L. Usefulness of a New Large Set of High Throughput EST-SNP Markers as a Tool for Olive Germplasm Collection Management. Front. Plant Sci. 2018, 9, 1320. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.A.; Churchill, G.A.; Autroque, J.E.; Tanksley, S.D.; Swells, M.E. Optimising selection for plant linkage map. Genome 1993, 36, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Eltaher, S.; Sallam, A.; Belamkar, V.; Emara, H.A.; Nower, A.; Salem, K.; Poland, J.; Baenziger, P.S. Genetic Diversity and Population Structure of F3:6 Nebraska Winter Wheat Genotypes Using Genotyping-By-Sequencing. Front. Genet. 2018, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Belaj, A.; del Carmen Dominguez-García, M.; Atienza, S.G.; Urdíroz, N.M.; De la Rosa, R.; Satovic, Z.; Martín, A.; Kilian, A.; Trujillo, I.; Valpuesta, V.; et al. Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genomes 2012, 8, 365–378. [Google Scholar] [CrossRef]
- El Bakkali, A.; Haouane, H.; Moukhli, A.; Costes, E.; Van Damme, P.; Khadari, B. Construction of Core Collections Suitable for Association Mapping to Optimize Use of Mediterranean Olive (Olea europaea L.) Genetic Resources. PLoS ONE 2013, 8, e61265. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | No. of Genotypes | |
|---|---|---|
| Climatic zone | Country | |
| Continental | Russia | 2 |
| Dry | Algeria | 1 |
| Argentina | 1 | |
| Chile | 1 | |
| Egypt | 3 | |
| Israel | 1 | |
| Morocco | 1 | |
| Pakistan | 1 | |
| Palestine | 1 | |
| Syria | 1 | |
| Tunisia | 5 | |
| Temperate | Albania | 2 |
| Cyprus | 3 | |
| France | 5 | |
| Greece | 5 | |
| Italy | 22 | |
| Japan | 1 | |
| Spain | 14 | |
| US | 17 | |
| Tropical | Colombia | 1 |
| Peru | 2 | |
| Unknown | 6 | |
| Transitions | Transversions | |||||
|---|---|---|---|---|---|---|
| Type of SNP | A/G | C/T | A/C | A/T | G/T | C/G |
| Number of sites | 15,875 | 16,010 | 5513 | 7476 | 5288 | 3913 |
| Frequencies | 29.36% | 29.61% | 10.20% | 13.83% | 9.78% | 7.24% |
| Total | 31,885 (59.96%) | 22,190 (41.04%) | ||||
| Source of Variation | df | SS | MS | Variation | p-Value |
|---|---|---|---|---|---|
| Between the population | 7 | 630,346.08 | 90,049.44 | 32.94 | 0.001 |
| Within the population | 88 | 1,277,390.84 | 14,515.80 | 67.06 | 0.001 |
| Total | 95 | 1,907,736.91 | 20,081.44 | 100.00 |
| Source of Variation | df | SS | MS | Variation | p-Value |
|---|---|---|---|---|---|
| Between the population | 7 | 310,271.04 | 44,324.43 | 4.29 | 0.04 |
| Between the climatic zones within the population | 13 | 493,233.74 | 37,941.06 | 25.82 | 0.001 |
| Within the population | 75 | 1,104,232.13 | 14,723.10 | 69.89 | 0.001 |
| Total | 95 | 1,907,736.91 | 20,081.44 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, A.S.M.F.; Sanders, D.; Mishra, A.K.; Joshi, V. Genetic Diversity and Population Structure Analysis of the USDA Olive Germplasm Using Genotyping-By-Sequencing (GBS). Genes 2021, 12, 2007. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122007
Islam ASMF, Sanders D, Mishra AK, Joshi V. Genetic Diversity and Population Structure Analysis of the USDA Olive Germplasm Using Genotyping-By-Sequencing (GBS). Genes. 2021; 12(12):2007. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122007
Chicago/Turabian StyleIslam, A. S. M. Faridul, Dean Sanders, Amit Kumar Mishra, and Vijay Joshi. 2021. "Genetic Diversity and Population Structure Analysis of the USDA Olive Germplasm Using Genotyping-By-Sequencing (GBS)" Genes 12, no. 12: 2007. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122007