
SUMO-Based Regulation of Nuclear Positioning to Spatially Regulate Homologous Recombination Activities at Replication Stress Sites


Abstract
:1. Replication Stressed Forks and Homologous Recombination
2. Replication Stress Sites Move to the Nuclear Periphery
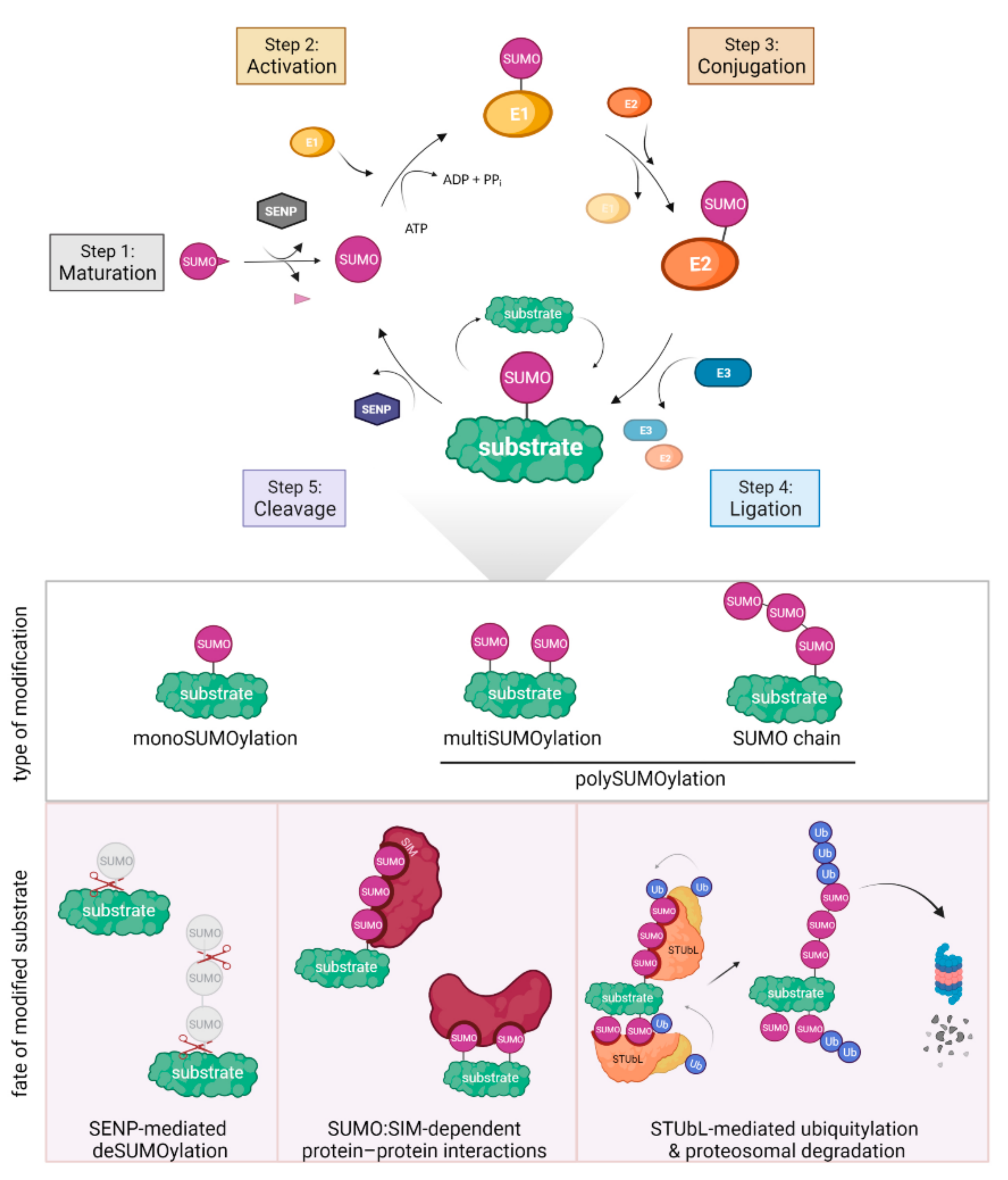
3. SUMOylation in DNA Repair
4. NPCs Anchor DNA Lesions in a SUMO-Dependent Manner to Promote DNA Repair
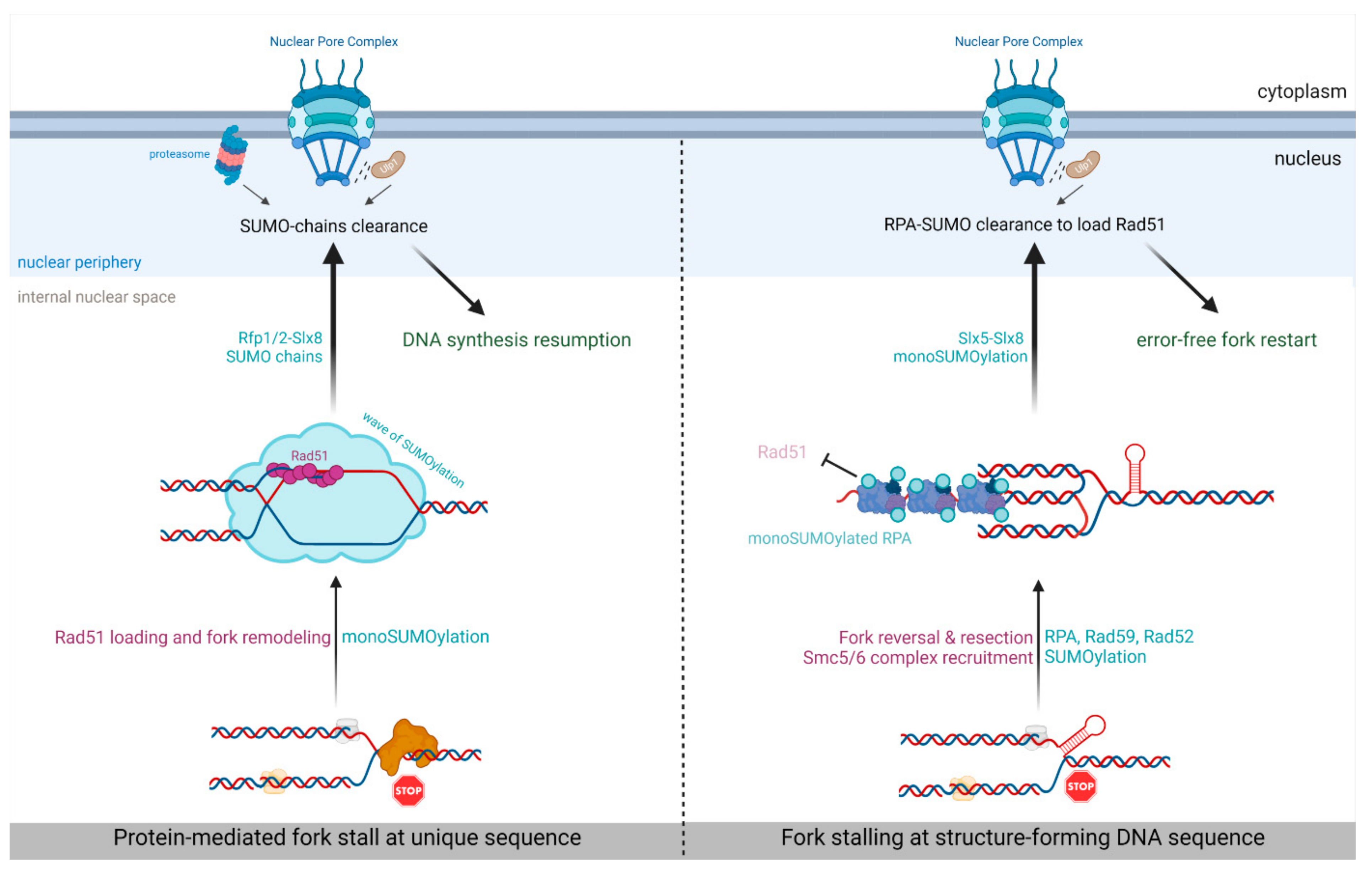
5. SUMO-Based Regulation of Nuclear Positioning Regulates Replication Fork Repair
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willaume, S.; Rass, E.; Paulafontanilla-Ramirez, P.; Moussa, A.; Wanschoor, P.; Bertrand, P. A link between replicative stress, lamin proteins, and inflammation. Genes 2021, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A.E. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Pasero, P. Replication stress: From chromatin to immunity and beyond. Curr. Opin. Genet. Dev. 2021, 71, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–280. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Elango, R.; Panday, A.; Willis, N.A. Recombination and restart at blocked replication forks. Curr. Opin. Genet. Dev. 2021, 71, 154–162. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Puddu, F.; Costanzo, V. RAD51-and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2012, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Lambert, S. Recombination-dependent replication: New perspectives from site-specific fork barriers. Curr. Opin. Genet. Dev. 2021, 71, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [Green Version]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef]
- Teixeira-Silva, A.; Ait Saada, A.; Hardy, J.; Iraqui, I.; Nocente, M.C.; Fréon, K.; Lambert, S.A.E. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalousi, A.; Soutoglou, E. Nuclear compartmentalization of DNA repair. Curr. Opin. Genet. Dev. 2016, 37, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; Van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA Double-Strand Breaks Revealed by Clustering of Damaged Chromosome Domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeber, A.; Gasser, S.M. Chromatin organization and dynamics in double-strand break repair. Curr. Opin. Genet. Dev. 2017, 43, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, S.; Dubrana, K.; Tsai-Pflugfelder, M.; Davidson, M.B.; Roberts, T.M.; Brown, G.W.; Varela, E.; Hediger, F.; Gasser, S.M.; Krogan, N.J. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 2008, 322, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horigome, C.; Unozawa, E.; Ooki, T.; Kobayashi, T. Ribosomal RNA gene repeats associate with the nuclear pore complex for maintenance after DNA damage. PLoS Genet. 2019, 15, e1008103. [Google Scholar] [CrossRef]
- Marnef, A.; Finoux, A.L.; Arnould, C.; Guillou, E.; Daburon, V.; Rocher, V.; Mangeat, T.; Mangeot, P.E.; Ricci, E.P.; Legube, G. A cohesin/HUSH- And LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 2019, 33, 1175–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear position dictates DNA repair pathway choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [Green Version]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell 2009, 33, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Oza, P.; Jaspersen, S.L.; Miele, A.; Dekker, J.; Peterson, C.L. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009, 23, 912–927. [Google Scholar] [CrossRef] [Green Version]
- Horigome, C.; Oma, Y.; Konishi, T.; Schmid, R.; Marcomini, I.; Hauer, M.H.; Dion, V.; Harata, M.; Gasser, S.M. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol. Cell 2014, 55, 626–639. [Google Scholar] [CrossRef] [Green Version]
- Khadaroo, B.; Teixeira, M.T.; Luciano, P.; Eckert-Boulet, N.; Germann, S.M.; Simon, M.N.; Gallina, I.; Abdallah, P.; Gilson, E.; Géli, V.; et al. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009, 11, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Churikov, D.; Charifi, F.; Eckert-Boulet, N.; Silva, S.; Simon, M.-N.; Lisby, M.; Géli, V. SUMO-Dependent Relocalization of Eroded Telomeres to Nuclear Pore Complexes Controls Telomere Recombination. Cell Rep. 2016, 15, 1242–1253. [Google Scholar] [CrossRef] [Green Version]
- Su, X.A.; Dion, V.; Gasser, S.M.; Freudenreich, C.H. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 2015, 29, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramarz, K.; Schirmeisen, K.; Boucherit, V.; Ait Saada, A.; Lovo, C.; Palancade, B.; Freudenreich, C.; Lambert, S.A.E. The nuclear pore primes recombination-dependent DNA synthesis at arrested forks by promoting SUMO removal. Nat. Commun. 2020, 11, 5643. [Google Scholar] [CrossRef]
- Aguilera, P.; Whalen, J.; Minguet, C.; Churikov, D.; Freudenreich, C.; Simon, M.N.; Géli, V. The nuclear pore complex prevents sister chromatid recombination during replicative senescence. Nat. Commun. 2020, 11, 160. [Google Scholar] [CrossRef] [Green Version]
- Lamm, N.; Read, M.N.; Nobis, M.; Van Ly, D.; Page, S.G.; Masamsetti, V.P.; Timpson, P.; Biro, M.; Cesare, A.J. Nuclear F-actin counteracts nuclear deformation and promotes fork repair during replication stress. Nat. Cell Biol. 2020, 22, 1460–1470. [Google Scholar] [CrossRef]
- Pinzaru, A.M.; Kareh, M.; Lamm, N.; Lazzerini-Denchi, E.; Cesare, A.J.; Sfeir, A. Replication stress conferred by POT1 dysfunction promotes telomere relocalization to the nuclear pore. Genes Dev. 2020, 34, 1619–1636. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Rogers, S.; Cesare, A.J. Chromatin mobility and relocation in DNA repair. Trends Cell Biol. 2021, 31, 843–855. [Google Scholar] [CrossRef]
- Horigome, C.; Bustard, D.E.; Marcomini, I.; Delgoshaie, N.; Tsai-Pflugfelder, M.; Cobb, J.A.; Gasser, S.M. PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev. 2016, 30, 931–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Fréon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef]
- Ait Saada, A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Fréon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398–410.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, E.; Miyabe, I.; Iraqui, I.; Zheng, J.; Lambert, S.A.E.; Carr, A.M. The extent of error-prone replication restart by homologous recombination is controlled by Exo1 and checkpoint proteins. J. Cell Sci. 2014, 127, 2983–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyabe, I.; Mizuno, K.; Keszthelyi, A.; Daigaku, Y.; Skouteri, M.; Mohebi, S.; Kunkel, T.A.; Murray, J.M.; Carr, A.M. Polymerase δ replicates both strands after homologous recombination–dependent fork restart. Nat. Struct. Mol. Biol. 2015, 22, 932–938. [Google Scholar] [CrossRef]
- Nguyen, M.O.; Jalan, M.; Morrow, C.A.; Osman, F.; Whitby, M.C. Recombination occurs within minutes of replication blockage by RTS1 producing restarted forks that are prone to collapse. Elife 2015, 4, e04539. [Google Scholar] [CrossRef]
- Pichler, A.; Fatouros, C.; Lee, H.; Eisenhardt, N. SUMO conjugation—A mechanistic view. Biomol. Concepts 2017, 8, 13–36. [Google Scholar] [CrossRef]
- Ulrich, H.D. The Fast-Growing Business of SUMO Chains. Mol. Cell 2008, 32, 301–305. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Meyer-Teschendorf, K.; Pabst, S.; Ulrich, H.D.; Gehring, N.H.; Hofmann, K.; Praefcke, G.J.K.; Dohmen, R.J. Arkadia/RNF111 is a SUMO-targeted ubiquitin ligase with preference for substrates marked with SUMO1-capped SUMO2/3 chain. Nat. Commun. 2019, 10, 3678. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lee, C.C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef] [PubMed]
- Watts, F.Z.; Skilton, A.; Ho, J.C.Y.; Boyd, L.K.; Trickey, M.A.M.; Gardner, L.; Ogi, F.X.; Outwin, E.A. The role of Schizosaccharomyces pombe SUMO ligases in genome stability. In Proceedings of the Biochemical Society Transactions. Biochem. Soc. Trans. 2007, 35, 1379–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacher, M.; Pfander, B.; Jentsch, S. Identification of SUMO-protein conjugates. Methods Enzymol. 2005, 399, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, S.; Psakhye, I. Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu. Rev. Genet. 2013, 47, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Soria-Bretones, I.; Cepeda-García, C.; Checa-Rodriguez, C.; Heyer, V.; Reina-San-Martin, B.; Soutoglou, E.; Huertas, P. DNA end resection requires constitutive sumoylation of CtIP by CBX4. Nat. Commun. 2017, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.J.; Hossain, L.; McCrostie, G.; Ronato, D.A.; Fitieh, A.; Rafique, T.A.; Mashayekhi, F.; Motamedi, M.; Masson, J.Y.; Ismail, I.H. SUMOylation mediates CtIP’s functions in DNA end resection and replication fork protection. Nucleic Acids Res. 2021, 49, 928–953. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Muñoz, J.; et al. A Proteomic Characterization of Factors Enriched at Nascent DNA Molecules. Cell Rep. 2013, 3, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Cremona, C.A.; Sarangi, P.; Yang, Y.; Hang, L.E.; Rahman, S.; Zhao, X. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol. Cell 2012, 45, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to sumo modifications in response to heat shock. Sci. Signal. 2009, 2, ra24. [Google Scholar] [CrossRef] [Green Version]
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and Mms21-Mediated Sumoylation Counteracts Recombinogenic Events at Damaged Replication Forks. Cell 2006, 127, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.L. A SUMOry of DNA Replication: Synthesis, Damage, and Repair. Cell 2006, 127, 455–457. [Google Scholar] [CrossRef] [Green Version]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Sarangi, P.; Zhao, X. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci. 2015, 40, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiten-Schmitz, J.; Schunck, K.; Müller, S. SUMO Chains Rule on Chromatin Occupancy. Front. Cell Dev. Biol. 2020, 7, 343. [Google Scholar] [CrossRef] [PubMed]
- Prudden, J.; Perry, J.J.P.; Nie, M.; Vashisht, A.A.; Arvai, A.S.; Hitomi, C.; Guenther, G.; Wohlschlegel, J.A.; Tainer, J.A.; Boddy, M.N. DNA Repair and Global Sumoylation Are Regulated by Distinct Ubc9 Noncovalent Complexes. Mol. Cell. Biol. 2011, 31, 2299–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Reindle, A.; Belichenko, I.; Bylebyl, G.R.; Chen, X.L.; Gandhi, N.; Johnson, E.S. Multiple domains in Siz SUMO ligases contribute to substrate selectivity. J. Cell Sci. 2006, 119, 4749–4757. [Google Scholar] [CrossRef] [Green Version]
- Xhemalce, B.; Riising, E.M.; Baumann, P.; Dejean, A.; Arcangioli, B.; Seeler, J.S. Role of SUMO in the dynamics of telomere maintenance in fission yeast. Proc. Natl. Acad. Sci. USA 2007, 104, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Pichler, A.; Gast, A.; Seeler, J.S.; Dejean, A.; Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 2002, 108, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Tang, J.; Xu, C.; Zhao, H.; Zhou, Y.; Wang, Y.; Yang, M.; Chen, X.; Chen, J. Histone deacetylase 4 inhibits NF-κB activation by facilitating IκBα sumoylation. J. Mol. Cell Biol. 2020, 12, 933–945. [Google Scholar] [CrossRef]
- Peng, J.; Wysocka, J. It takes a PHD to SUMO. Trends Biochem. Sci. 2008, 33, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Melhuish, T.A.; Wotton, D. The polycomb protein Pc2 is a SUMO E3. Cell 2003, 113, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Weger, S.; Hammer, E.; Heilbronn, R. Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Lett. 2005, 579, 5007–5012. [Google Scholar] [CrossRef] [Green Version]
- Palancade, B.; Liu, X.; Garcia-Rubio, M.; Aguilera, A.; Zhao, X.; Doye, V. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol. Biol. Cell 2007, 18, 2912–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, K.; Piller, T.; Müller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131, jcs211904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palancade, B.; Doye, V. Sumoylating and desumoylating enzymes at nuclear pores: Underpinning their unexpected duties? Trends Cell Biol. 2008, 18, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Uzunova, K.; Göttsche, K.; Miteva, M.; Weisshaar, S.R.; Glanemann, C.; Schnellhardt, M.; Niessen, M.; Scheel, H.; Hofmann, K.; Johnson, E.S.; et al. Ubiquitin-dependent proteolytic control of SUMO conjugates. J. Biol. Chem. 2007, 282, 34167–34175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Oram, M.K.; Bielinsky, A.K. Sumo-targeted ubiquitin ligases and their functions in maintaining genome stability. Int. J. Mol. Sci. 2021, 22, 5391. [Google Scholar] [CrossRef]
- Kramarz, K.; Litwin, I.; Cal-Bakowska, M.; Szakal, B.; Branzei, D.; Wysocki, R.; Dziadkowiec, D. Swi2/Snf2-like protein Uls1 functions in the Sgs1-dependent pathway of maintenance of rDNA stability and alleviation of replication stress. DNA Repair 2014, 21, 24–35. [Google Scholar] [CrossRef]
- Prudden, J.; Pebernard, S.; Raffa, G.; Slavin, D.A.; Perry, J.J.P.; Tainer, J.A.; McGowan, C.H.; Boddy, M.N. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 2007, 26, 4089–4101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psakhye, I.; Castellucci, F.; Branzei, D. SUMO-Chain-Regulated Proteasomal Degradation Timing Exemplified in DNA Replication Initiation. Mol. Cell 2019, 76, 632–645.e6. [Google Scholar] [CrossRef] [Green Version]
- Liebelt, F.; Jansen, N.S.; Kumar, S.; Gracheva, E.; Claessens, L.A.; Verlaan-de Vries, M.; Willemstein, E.; Vertegaal, A.C.O. The poly-SUMO2/3 protease SENP6 enables assembly of the constitutive centromere-associated network by group deSUMOylation. Nat. Commun. 2019, 10, 3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, J.R.; Das, M.; Brill, S.J. Genetic evidence that polysumoylation bypasses the need for a SUMO-targeted UB ligase. Genetics 2011, 187, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Fernandez-Martinez, J.; Nudelman, I.; Shi, Y.; Zhang, W.; Raveh, B.; Herricks, T.; Slaughter, B.D.; Hogan, J.A.; Upla, P.; et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature 2018, 555, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Appen, A.; Beck, M. Structure Determination of the Nuclear Pore Complex with Three-Dimensional Cryo electron Microscopy. J. Mol. Biol. 2016, 428, 2001–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knockenhauer, K.E.; Schwartz, T.U. The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell 2016, 164, 1162–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelz, A.; Debler, E.W.; Blobel, G. The Structure of the nuclear pore complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, T.U. The Structure Inventory of the Nuclear Pore Complex. J. Mol. Biol. 2016, 428, 1986–2000. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, A.; Hetzer, M.W. Nuclear pore proteins and the control of genome functions. Genes Dev. 2015, 29, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, M.A.; Hetzer, M.W. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 2008, 18, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whalen, J.M.; Freudenreich, C.H. Location, location, location: The role of nuclear positioning in the repair of collapsed forks and protection of genome stability. Genes 2020, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Loeillet, S.; Palancade, B.; Cartron, M.; Thierry, A.; Richard, G.-F.; Dujon, B.; Doye, V.; Nicolas, A. Genetic network interactions among replication, repair and nuclear pore deficiencies in yeast. DNA Repair 2005, 4, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Moudry, P.; Lukas, C.; Macurek, L.; Neumann, B.; Heriche, J.K.; Pepperkok, R.; Ellenberg, J.; Hodny, Z.; Lukas, J.; Bartek, J. Nucleoporin NUP153 guards genome integrity by promoting nuclear import of 53BP1. Cell Death Differ. 2012, 19, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaître, C.; Fischer, B.; Kalousi, A.; Hoffbeck, A.S.; Guirouilh-Barbat, J.; Shahar, O.D.; Genet, D.; Goldberg, M.; Betrand, P.; Lopez, B.; et al. The nucleoporin 153, a novel factor in double-strand break repair and DNA damage response. Oncogene 2012, 31, 4803–4809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; Santos-Pereira, J.M.; Aguilera, A. The Nup84 complex coordinates the DNA damage response to warrant genome integrity. Nucleic Acids Res. 2019, 47, 4054–4067. [Google Scholar] [CrossRef] [PubMed]
- Amaral, N.; Ryu, T.; Li, X.; Chiolo, I. Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 2017, 33, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Whalen, J.M.; Dhingra, N.; Wei, L.; Zhao, X.; Freudenreich, C.H. Relocation of Collapsed Forks to the Nuclear Pore Complex Depends on Sumoylation of DNA Repair Proteins and Permits Rad51 Association. Cell Rep. 2020, 31, 107635. [Google Scholar] [CrossRef] [PubMed]
- Capella, M.; Mandemaker, I.K.; Martín Caballero, L.; den Brave, F.; Pfander, B.; Ladurner, A.G.; Jentsch, S.; Braun, S. Nucleolar release of rDNA repeats for repair involves SUMO-mediated untethering by the Cdc48/p97 segregase. Nat. Commun. 2021, 12, 4918. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; Sunjevaric, I.; De Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragón, L.; Lisby, M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Géli, V.; Lisby, M. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays 2015, 37, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Polleys, E.J.; Freudenreich, C.H. Homologous recombination within repetitive DNA. Curr. Opin. Genet. Dev. 2021, 71, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Zhao, X. DNA break-induced sumoylation is enabled by collaboration between a sumo ligase and the ssdnabinding complex rpa. Genes Dev. 2015, 29, 1593–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, M.; Boddy, M.N. Pli1 PIAS1 SUMO Ligase Protected by the Nuclear Pore-associated SUMO Protease Ulp1 SENP1/2. J. Biol. Chem. 2015, 290, 22678–22685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshidari, R.; Huang, R.; Medghalchi, M.; Tse, E.Y.W.; Ashgriz, N.; Lee, H.O.; Wyatt, H.; Mekhail, K. DNA repair by Rad52 liquid droplets. Nat. Commun. 2020, 11, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| SUMO Pathway Component | Humans | S. cerevisiae | S. pombe | |
|---|---|---|---|---|
| Small ubiquitin-like modifier (SUMO) | SUMO-1, SUMO-2, SUMO-3, SUMO-4, SUMO-5 | Smt3 | Pmt3 | |
| Activating enzyme (E1) | SAE1 SAE2 | Aos1 Uba2 | Rad31 Fub2 | |
| Conjugating enzyme (E2) | Ubc9 | Ubc9 | Hus5 | |
| SUMO ligase (E3) | SP-RING type | PIAS1, PIAS2, PIAS3, PIAS4 Mms21 | Siz1, Siz2 Mms21 Zip3 | Pli1Nse2 |
| other | RanBP2 * [69] HDAC4 [70], KPA1 [71], Pc2 [72], Topors [73] | |||
| SUMO-targeted ubiquitin ligase (STUbL) | RNF4 RNF11 | Slx5-Slx8 Uls1 | Rfp1/Rfp2-Slx8 Rrp2 (predicted) | |
| Sentrin/SUMO-specific protease (SENP) | SENP1 °,*, SENP2 °,*, SENP3, SENP5 ° SENP6, SENP7 | Ulp1 °,* Ulp2 | Ulp1 °,* Ulp2 | |
| Type of Obstacle | Protein-Mediated Fork Arrest | Structure-Forming DNA Sequence | Telomere-Specific Replication Stress | Aphidicolin Induced Replication Stress | |
|---|---|---|---|---|---|
| System description | Site-specific RFB blocking a single replisome in a polar manner | Expanded trinucleotide repeats forming hairpin structures that stall replisomes | Stalled replisomes at telomere repeats in telomerase-negative cells | Telomere-specific replication stress induced by POT1 dysfunctions | Global replication fork stalling induced |
| Organism | S. pombe | S. cerevisiae | S. cerevisiae | human cell lines | human cell lines |
| Relocation and anchorage requirements | ● Rad51-dependent fork remodeling ● Pli1 ● SUMO chain ● Rfp1-Slx8, ● Rfp2-Slx8 ● NPC-anchorage site unknown | ● Nascent DNA degradation (by Mre11, Exo1, Dna2) ● Mms21 ● SUMOylation of RPA, Rad52, Rad59 ● Slx5-SUMO interaction ● Nup1, Nup84 | ● Nup1 | ● F-actin polymerization ● ATR pathway ● Nup62, Nup153, TPR | ● F-actin polymerization |
| Relocation outcomes | Ulp1-NPCs alleviate inhibitory effect of SUMO chains on HR-mediated fork restart | Rad51 loading to promote error free fork restart and preventing CAG repeat instability | Promoting conservative fork restart pathway to avoid error-prone Rad51-dependent SCR | Preventing SCR at telomeres to promote the maintenance of repetitive DNA | Promoting replication stress response to ensure fork restart and prevent mitotic abnormalities. |
| Reference | [33] | [32,34,97] | [34] | [36] | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirmeisen, K.; Lambert, S.A.E.; Kramarz, K. SUMO-Based Regulation of Nuclear Positioning to Spatially Regulate Homologous Recombination Activities at Replication Stress Sites. Genes 2021, 12, 2010. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122010
Schirmeisen K, Lambert SAE, Kramarz K. SUMO-Based Regulation of Nuclear Positioning to Spatially Regulate Homologous Recombination Activities at Replication Stress Sites. Genes. 2021; 12(12):2010. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122010
Chicago/Turabian StyleSchirmeisen, Kamila, Sarah A. E. Lambert, and Karol Kramarz. 2021. "SUMO-Based Regulation of Nuclear Positioning to Spatially Regulate Homologous Recombination Activities at Replication Stress Sites" Genes 12, no. 12: 2010. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122010