
Exome Sequencing Reveals Novel Variants and Expands the Genetic Landscape for Congenital Microcephaly

 , , , , , ,
, , , , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Characteristics of the Cohort
3.2. Definitive and Likely Diagnosis Cases
3.3. Dual Molecular Diagnosis
3.4. Novel Gene Associations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woods, C.G. Human microcephaly. Curr. Opin. Neurobiol. 2004, 14, 112–117. [Google Scholar] [CrossRef]
- Von Der Hagen, M.; Pivarcsi, M.; Liebe, J.; von Bernuth, H.; Di Donato, N.; Hennermann, J.B.; Bührer, C.; Wieczorek, D.; Kaindl, A.M. Diagnostic approach to microcephaly in childhood: A two-center study and review of the literature. Dev. Med. Child Neurol. 2014, 56, 732–741. [Google Scholar] [CrossRef]
- Woods, C.G.; Parker, A. Investigating microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.N.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genom. 2015, 8 (Suppl. S1), S4. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, L.E.; Paciorkowski, A.R. Genetic disorders associated with postnatal microcephaly. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, D.; O’Driscoll, M. Congenital microcephaly. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166, 124–139. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [Green Version]
- Barbelanne, M.; Tsang, W.Y. Molecular and Cellular Basis of Autosomal Recessive Primary Microcephaly. BioMed Res. Int. 2014, 2014, 547986. [Google Scholar] [CrossRef]
- Perez, Y.; Bar-Yaacov, R.; Kadir, R.; Wormser, O.; Shelef, I.; Birk, O.; Flusser, H.; Birnbaum, R.Y. Mutations in the microtubule-associated protein MAP11 (C7orf43) cause microcephaly in humans and zebrafish. Brain 2019, 142, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Rump, P.; Jazayeri, O.; van Dijk-Bos, K.K.; Johansson, L.F.; van Essen, A.J.; Verheij, J.B.G.M.; Veenstra-Knol, H.E.; Redeker, E.J.W.; Mannens, M.M.A.M.; Swertz, M.A.; et al. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med. Genom. 2015, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Boonsawat, P.; Joset, P.; Steindl, K.; Oneda, B.; Gogoll, L.; Azzarello-Burri, S.; Sheth, F.; Datar, C.; Verma, I.C.; Puri, R.D.; et al. Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genet. Med. 2019, 21, 2043–2058. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Maddirevula, S.; Ewida, N.; Alsahli, S.; Abdel-Salam, G.M.H.; Zaki, M.S.; Al Tala, S.; Alhashem, A.; Softah, A.; Al-Owain, M.; et al. Genomic and phenotypic delineation of congenital microcephaly. Genet. Med. 2018, 21, 545–552. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017, 201178. [Google Scholar] [CrossRef] [Green Version]
- Chapman, B.; Kirchner, R.; Pantano, L.; Khotiainsteva, T.; Smet, M.D.; Beltrame, L.; Saveliev, V.; Guimera, R.V.; Nau-menko, S.; Kern, J.; et al. Bcbio/Bcbio-Nextgen: V1.1.9; Zenodo: Geneva, Switzerland, 2019. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E.; Project, N.E.S. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick Coverage Calculation for Genomes and Exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 2014, 137, 1676–1700. [Google Scholar] [CrossRef] [Green Version]
- Moortgat, S.; Berland, S.; Aukrust, I.; Maystadt, I.; Baker, L.; Benoit, V.; Caro-Llopis, A.; Cooper, N.S.; Debray, F.-G.; Faivre, L.; et al. HUWE1 variants cause dominant X-linked intellectual disability: A clinical study of 21 patients. Eur. J. Hum. Genet. 2018, 26, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvish, H.; Esmaeeli-Nieh, S.; Monajemi, G.B.; Mohseni, M.; Ghasemi-Firouzabadi, S.; Abedini, S.S.; Bahman, I.; Jamali, P.; Azimi, S.; Mojahedi, F.; et al. A clinical and molecular genetic study of 112 Iranian families with primary microcephaly. J. Med. Genet. 2010, 47, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Gardella, E.; Møller, R. Phenotypic and genetic spectrum of SCN 8A -related disorders, treatment options, and outcomes. Epilepsia 2019, 60, S77–S85. [Google Scholar] [CrossRef] [Green Version]
- Zarate, Y.A.; Bosanko, K.A.; Caffrey, A.R.; Bernstein, J.A.; Martin, D.M.; Williams, M.S.; Berry-Kravis, E.M.; Mark, P.; Manning, M.A.; Bhambhani, V.; et al. Mutation update for the SATB2 gene. Hum. Mutat. 2019, 40, 1013–1029. [Google Scholar] [CrossRef] [Green Version]
- Borlot, F.; Abushama, A.; Morrison-Levy, N.; Jain, P.; Vinayan, K.P.; Abukhalid, M.; Aldhalaan, H.M.; Almuzaini, H.S.; Gulati, S.; Hershkovitz, T.; et al. KCNT1-related epilepsy: An international multicenter cohort of 27 pediatric cases. Epilepsia 2020, 61, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Lemke, J.R. GRIN1-Related Neurodevelopmental Disorder. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Ke, Z.; Chen, Y. Case Report: A de novo CTNNB1 Nonsense Mutation Associated with Neurodevelopmental Disorder, Retinal Detachment, Polydactyly. Front. Pediatr. 2020, 8, 850. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.Z.; Bekheirnia, M.R.; Lewis, A.M.; Mefford, H.C.; Golden-Grant, K.; Tarczy-Hornoch, K.; Briere, L.C.; Sweetser, D.A.; Walker, M.A.; Kravets, E.; et al. Missense variants in CTNNB1 can be associated with vitreoretinopathy—Seven new cases of CTNNB1 -associated neurodevelopmental disorder including a previously unreported retinal phenotype. Mol. Genet. Genom. Med. 2021, 9, e1542. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.; Egger, J.I.; E Jongbloed, R.; van Putten, M.M.; Zandwijk, M.D.B.-V.; Zwemer, A.-S.; Pfundt, R.; Willemsen, M.H. A de novo CTNNB1 novel splice variant in an adult female with severe intellectual disability. Int. Med. Case Rep. J. 2020, 13, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Mitter, D.; Pringsheim, M.; Kaulisch, M.; Plümacher, K.S.; Schröder, S.; Warthemann, R.; Jamra, R.A.; Baethmann, M.; Bast, T.; Büttel, H.-M.; et al. FOXG1 syndrome: Genotype–phenotype association in 83 patients with FOXG1 variants. Genet. Med. 2018, 20, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Vanstone, M.R.; Hartley, T.; Osmond, M.; Barrowman, N.; Allanson, J.; Baker, L.; Dabir, T.A.; Dipple, K.M.; Dobyns, W.; et al. Mandibulofacial Dysostosis with Microcephaly: Mutation and Database Update. Hum. Mutat. 2016, 37, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.K.; Otero, L.J.; Legris, M.; Brown, R.M. Syndrome of the Month Pyruvate Dehydrogenase Deficiency. Amino Acids 1994, 31, 875–879. [Google Scholar]
- Pavlu-Pereira, H.; Silva, M.J.; Florindo, C.; Sequeira, S.; Ferreira, A.C.; Duarte, S.; Rodrigues, A.L.; Janeiro, P.; Oliveira, A.; Gomes, D.; et al. Pyruvate dehydrogenase complex deficiency: Updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients. Orphanet J. Rare Dis. 2020, 15, 298. [Google Scholar] [CrossRef]
- Verloes, A.; Drunat, S.; Passemard, S. ASPM Primary Microcephaly. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Abdel-Hamid, M.S.; Ismail, M.F.; Darwish, H.A.; Effat, L.K.; Zaki, M.S.; Abdel-Salam, G. Molecular and phenotypic spectrum of ASPM-related primary microcephaly: Identification of eight novel mutations. Am. J. Med. Genet. Part A 2016, 170, 2133–2140. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Akdemir, Z.H.C.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Lemke, J.R.; Geider, K.; Helbig, K.L.; Heyne, H.O.; Schütz, H.; Hentschel, J.; Courage, C.; Depienne, C.; Nava, C.; Heron, D.; et al. Delineating the GRIN1 phenotypic spectrum. Neurology 2016, 86, 2171–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Liu, B.; Yang, L.; Wang, H.; Wu, B.; Liu, R.; Chen, H.; Chen, X.; Yu, S.; Chen, B.; et al. Clinical exome sequencing as the first-tier test for diagnosing developmental disorders covering both CNV and SNV: A Chinese cohort. J. Med. Genet. 2020, 57, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Minczuk, M. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002, 30, 5074–5086. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, R.J.; Obriot, H.; Paczkowska, A.; Jedrzejczak, R.; Dmochowska, A.; Bartnik, E.; Formstecher, P.; Polakowska, R.; Stepien, P.P. Down-regulation of human RNA/DNA helicase SUV3 induces apoptosis by a caspase- and AIF-dependent pathway. Biol. Cell 2007, 99, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Doherty, K.M.; Brosh, R.M. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem. J. 2006, 398, 319–337. [Google Scholar] [CrossRef] [PubMed]
- De Renty, C.; Ellis, N.A. Bloom’s syndrome: Why not premature aging? Ageing Res. Rev. 2017, 33, 36–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouman, A.; van Koningsbruggen, S.; Karakullukcu, M.B.; Schreuder, W.H.; Lakeman, P. Bloom syndrome does not always present with sun-sensitive facial erythema. Eur. J. Med. Genet. 2018, 61, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.T.; Lu, J.; Zhu, M.; Maddison, L.A.; Boyd, K.L.; Huskey, L.; Ju, B.; Hesselson, D.; Zhong, T.P.; Page-McCaw, P.; et al. Conditional control of gene function by an invertible gene trap in zebrafish. Proc. Natl. Acad. Sci. USA 2012, 109, 15389–15394. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.; Mason, P.; Szczesny, R.; Maddukuri, L.; Dziwura, S.; Jedrzejczak, R.; Paul, E.; Wojcik, A.; Dybczynska, L.; Tudek, B. Interaction of human SUV3 RNA/DNA helicase with BLM helicase; loss of the SUV3 gene results in mouse embryonic lethality. Mech. Ageing Dev. 2007, 128, 609–617. [Google Scholar] [CrossRef]
- Lee, T.; Di Paola, D.; Malina, A.; Mills, J.R.; Kreps, A.; Grosse, F.; Tang, H.; Zannis-Hadjopoulos, M.; Larsson, O.; Pelletier, J. Suppression of the DHX9 Helicase Induces Premature Senescence in Human Diploid Fibroblasts in a p53-dependent Manner. J. Biol. Chem. 2014, 289, 22798–22814. [Google Scholar] [CrossRef] [Green Version]
- Capitanio, J.S.; Montpetit, B.; Wozniak, R.W. Human Nup98 regulates the localization and activity of DExH/D-box helicase DHX9. Elife 2017, 6, e18825. [Google Scholar] [CrossRef]
- Aktas, T.; Avsar Ilik, I.; Maticzka, D.; Bhardwaj, V.; Pessoa Rodrigues, C.; Mittler, G.; Manke, T.; Backofen, R.; Akhtar, A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 2017, 544, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Pelletier, J. The biology of DHX9 and its potential as a therapeutic target. Oncotarget 2016, 7, 42716–42739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, H.; Imai, T.; Imataka, H.; Tsujimoto, M.; Matsumoto, K.; Okano, H. Neural RNA-binding protein Musashi1 inhibits translation initiation by competing with eIF4G for PABP. J. Cell Biol. 2008, 181, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Cambuli, F.; Correa, B.; Rezza, A.; Burns, S.; Qiao, M.; Uren, P.; Kress, E.; Boussouar, A.; Galante, P.; Penalva, L.; et al. A Mouse Model of Targeted Musashi1 Expression in Whole Intestinal Epithelium Suggests Regulatory Roles in Cell Cycle and Stemness. St. Cells 2015, 33, 3621–3634. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.; Cieply, B.; Carstens, R.; Ramamurthy, V.; Stoilov, P. The Musashi 1 Controls the Splicing of Photoreceptor-Specific Exons in the Vertebrate Retina. PLoS Genet. 2016, 12, e1006256. [Google Scholar] [CrossRef]
- Abreu, R.D.S.; Sanchez-Diaz, P.C.; Vogel, C.; Burns, S.C.; Ko, D.; Burton, T.L.; Vo, D.T.; Chennasamudaram, S.; Le, S.-Y.; Shapiro, B.A.; et al. Genomic Analyses of Musashi1 Downstream Targets Show a Strong Association with Cancer-related Processes. J. Biol. Chem. 2009, 284, 12125–12135. [Google Scholar] [CrossRef] [Green Version]
- Siskos, N.; Stylianopoulou, E.; Skavdis, G.; Grigoriou, M. Molecular Genetics of Microcephaly Primary Hereditary: An Overview. Brain Sci. 2021, 11, 581. [Google Scholar] [CrossRef]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.D.; Coyaud, E.; Gonçalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [Green Version]
- Vandenbrouck, Y.; Pineau, C.; Lane, L. The Functionally Unannotated Proteome of Human Male Tissues: A Shared Resource to Uncover New Protein Functions Associated with Reproductive Biology. J. Proteome Res. 2020, 19, 4782–4794. [Google Scholar] [CrossRef]
- Kodani, A.; Yu, T.W.; Johnson, J.R.; Jayaraman, D.; Johnson, T.L.; Al-Gazali, L.; Sztriha, L.; Partlow, J.N.; Kim, H.; Krup, A.L.; et al. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife 2015, 4, e07519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomioka, N.H.; Yasuda, H.; Miyamoto, H.; Hatayama, M.; Morimura, N.; Matsumoto, Y.; Suzuki, T.; Odagawa, M.; Odaka, Y.S.; Iwayama, Y.; et al. Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nat. Commun. 2014, 5, 4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, J.; Mitchell, K.J. Mutation of Elfn1 in Mice Causes Seizures and Hyperactivity. PLoS ONE 2013, 8, e80491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Sarria, I.; Fehlhaber, K.E.; Kamasawa, N.; Orlandi, C.; James, K.N.; Hazen, J.L.; Gardner, M.R.; Farzan, M.; Lee, A.; et al. Mechanism for Selective Synaptic Wiring of Rod Photoreceptors into the Retinal Circuitry and Its Role in Vision. Neuron 2015, 87, 1248–1260. [Google Scholar] [CrossRef] [Green Version]



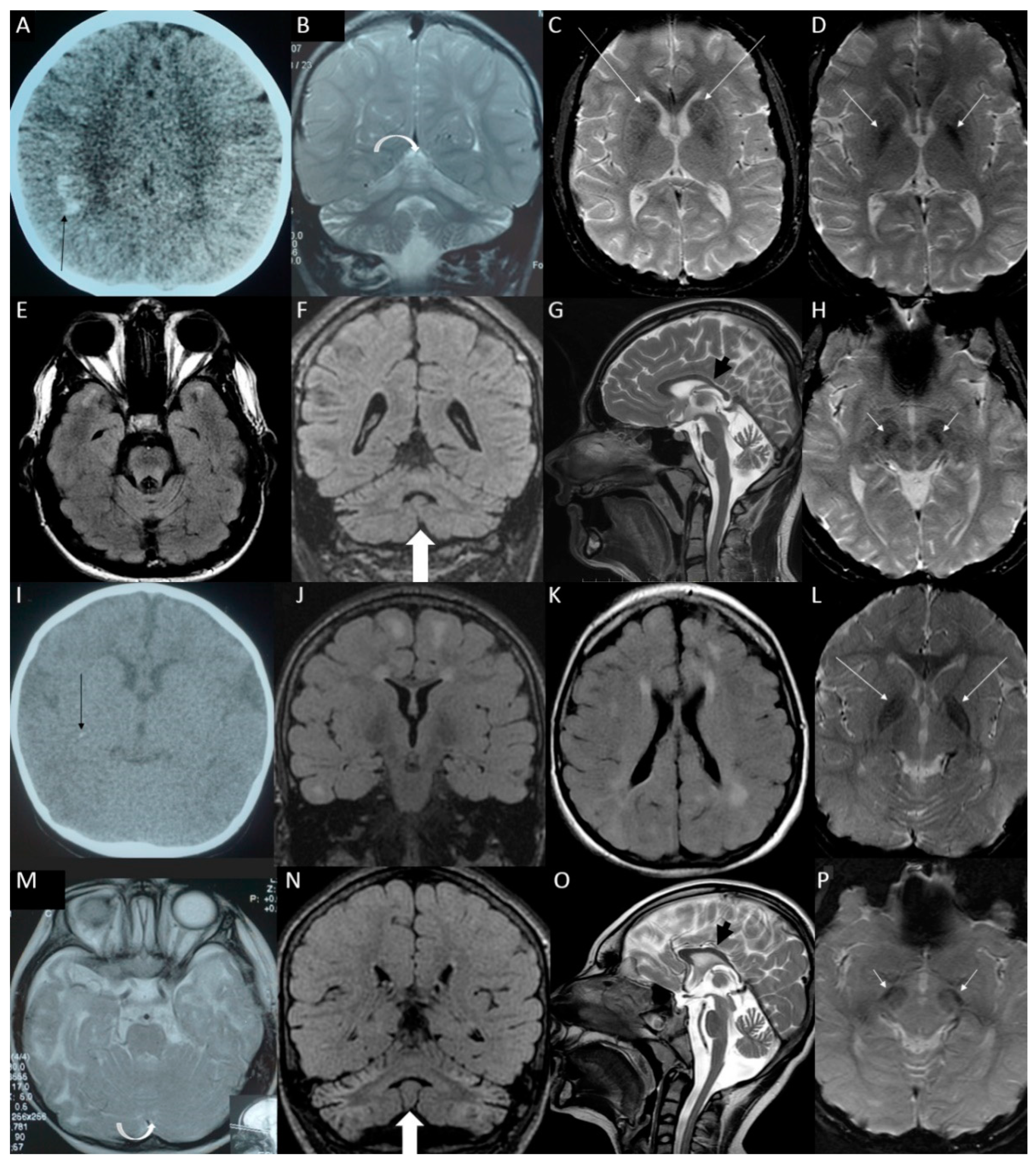{kind=link}
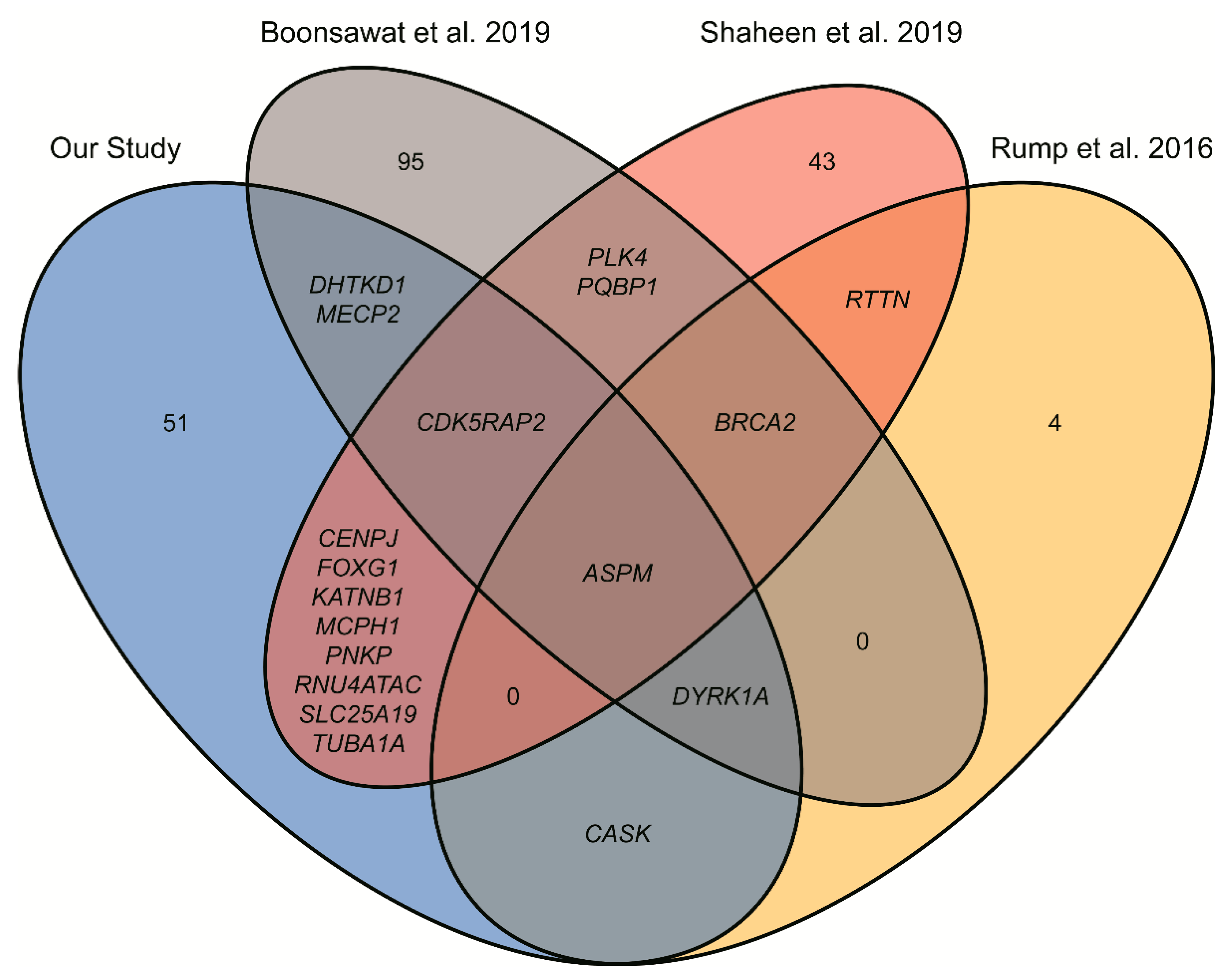{kind=link}
| Clinical Feature | HPO Number | Frequency |
|---|---|---|
| Microcephaly | HP:0000252 | 191 (100%) |
| Primary microcephaly | HP:0011451 | 77 (40.3%) |
| Secondary microcephaly | HP:0005484 | 110 (57.6%) |
| Unknown onset | HP:0000252 | 4 (2.1%) |
| Cognitive impairment (DD/ID) | HP:0100543 | 168 (87.9%) |
| Abnormal cerebral morphology | HP:0002060 | 133 (69.6%) |
| Abnormal corpus callosum morphology | HP:0001273 | 55 (28.8%) |
| Abnormal myelination | HP:0012447 | 35 (18.3%) |
| Abnormal cortical gyration | HP:0002536 | 28 (14.7%) |
| Abnormal cerebellum morphology | HP:0001317 | 28 (14.7%) |
| Ventriculomegaly | HP:0002119 | 22 (11.5%) |
| Abnormality of the nervous system | HP:0000707 | 166 (86.9%) |
| Abnormal muscle tone | HP:0003808 | 99 (51.8%) |
| Seizure | HP:0001250 | 76 (39.8%) |
| Refractory status epilepticus | HP:0032867 | 25 (13.1%) |
| Epileptic encephalopathy | HP:0200134 | 10 (5.2%) |
| Hemiplegia/hemiparesis or tetraplegia/tetraparesis | HP:0004374 HP:0030182 | 22 (11.5%) |
| Abnormality of movement | HP:0100022 | 17 (8.9%) |
| Stereotypy | HP:0000733 | 13 (6.8%) |
| Short stature | HP:0004322 | 61 (31.9%) |
| Abnormal facial shape | HP:0001999 | 75 (39.3%) |
| Strabismus | HP:0000486 | 27 (14.1%) |
| Abnormal heart morphology | HP:0001627 | 19 (9.9%) |
| Hearing impairment | HP:0000365 | 13 (6.8%) |
| Patient | Sex | Phenotype | Variant and Inheritance | Zygosity | OMIM Syndrome | OMIM ID | Syndrome Main Features | Pubmed |
|---|---|---|---|---|---|---|---|---|
| T50 | M | SM, ID, Dandy-Walker malformation, anterior commissure agenesis, dysmorphic facial features | TUBB2B-NM_178012.5 c.1171C>T p.(Arg391Cys) dn | het | Cortical dysplasia, complex, with other brain malformations 7 | 610031 | DD, polymicrogyria, corpus callosum agenesis, brainstem hypoplasia | [26] |
| HUWE1-NM_031407.7 c.11434G>A p.(Val3812Met) mat | hemi | Mental retardation, X-linked syndromic, Turner type | 309590 | DD, ID, hypotonia, speech delay, microcephaly, epilepsy, dysmorphic facial features | [27] | |||
| S78 | M | PM, DD, axial hypotonia, epileptic encephalopathy, limb hypertonia, EEG abnormalities | MCPH1-NM_024596.5 c.664T>C p.(Cys222Arg) | hom | Microcephaly 1, primary, autosomal recessive | 251200 | ID, microcephaly, short stature | [28] |
| SCN8A-NM_014191.4 c.5630A>G p.(Asn1877Ser) dn | het | Epileptic encephalopathy, early infantile, 13 | 614558 | DD, epilepsy, myoclonus, extrapyramidal signs | [29] | |||
| S177 | F | SM, DD, hypotonia, dysmorphic facial features | HUWE1-NM_031407.7 c.9208C>T p.(Arg3070Cys) dn | het | Mental retardation, X-linked syndromic, Turner type | 309590 | DD, ID, hypotonia, speech delay, microcephaly, epilepsy, dysmorphic facial features | [27] |
| SATB2-NM_015265.4 c.490G>A p.(Asp164Asn) dn | het | Glass syndrome | 612313 | DD, speech delay, dental anomalies, behavioural difficulties, feeding issues, abnormal brain neuroimaging, dysmorphic facial features | [30] | |||
| S188 | M | PM, DD, bilateral polymicrogyria, epilepsy, hypertonia, tetraplegia, nystagmus, strabismus convergent, cryptorchidism | KCNT1-NM_020822.3 c.1720G>A p.(Glu574Lys) dn | het | Epileptic encephalopathy, early infantile, 14 | 614959 | DD, epilepsy, absent speech, hypotonia, spasticity, microcephaly | [31] |
| GRIN1-NM_007327.4 c.1665G>T p.(Met555Ile) dn | het | Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant | 614254 | DD, epilepsy, hypotonia, absent speech, movement disorders, spasticity, visual impairment, bilateral polymicrogyria | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawidziuk, M.; Gambin, T.; Bukowska-Olech, E.; Antczak-Marach, D.; Badura-Stronka, M.; Buda, P.; Budzynska, E.; Castaneda, J.; Chilarska, T.; Czyzyk, E.; et al. Exome Sequencing Reveals Novel Variants and Expands the Genetic Landscape for Congenital Microcephaly. Genes 2021, 12, 2014. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122014
Dawidziuk M, Gambin T, Bukowska-Olech E, Antczak-Marach D, Badura-Stronka M, Buda P, Budzynska E, Castaneda J, Chilarska T, Czyzyk E, et al. Exome Sequencing Reveals Novel Variants and Expands the Genetic Landscape for Congenital Microcephaly. Genes. 2021; 12(12):2014. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122014
Chicago/Turabian StyleDawidziuk, Mateusz, Tomasz Gambin, Ewelina Bukowska-Olech, Dorota Antczak-Marach, Magdalena Badura-Stronka, Piotr Buda, Edyta Budzynska, Jennifer Castaneda, Tatiana Chilarska, Elzbieta Czyzyk, and et al. 2021. "Exome Sequencing Reveals Novel Variants and Expands the Genetic Landscape for Congenital Microcephaly" Genes 12, no. 12: 2014. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122014