
Gene Amplification as a Mechanism of Yeast Adaptation to Nonsense Mutations in Release Factor Genes

 , , , and
, , , and
Abstract
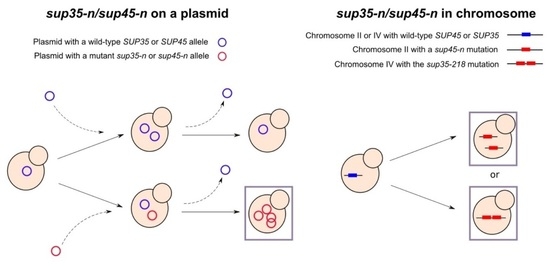
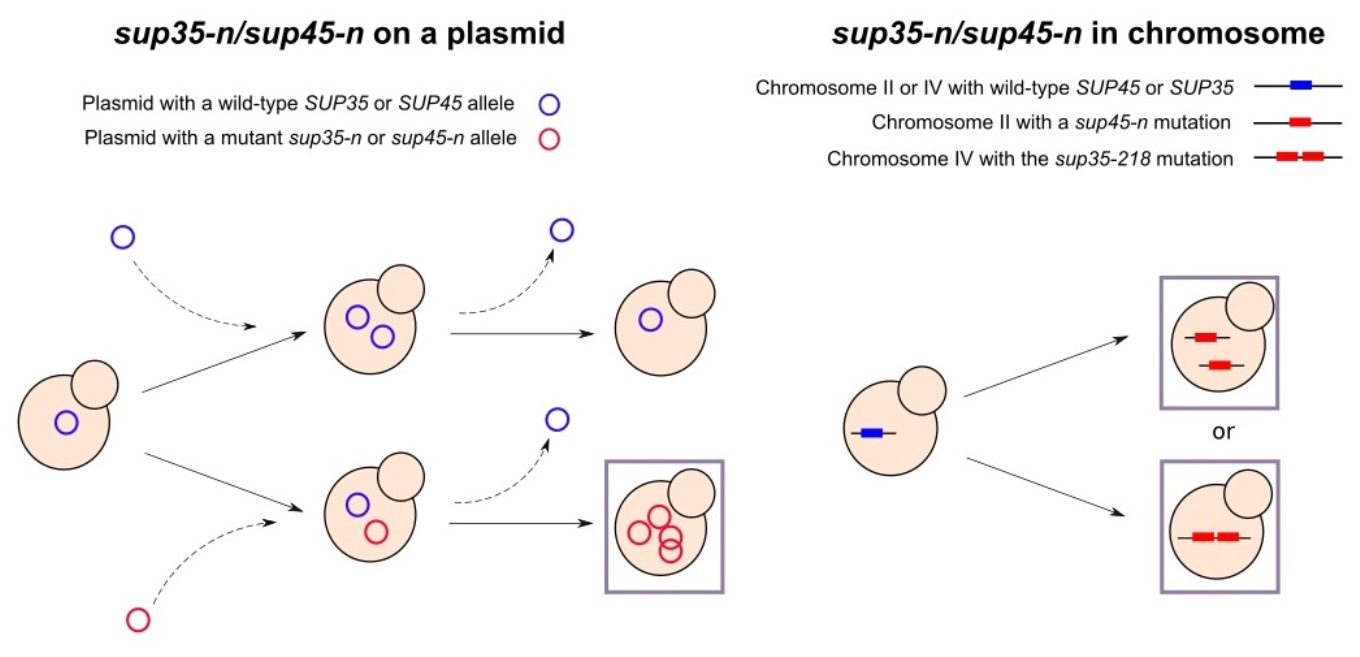
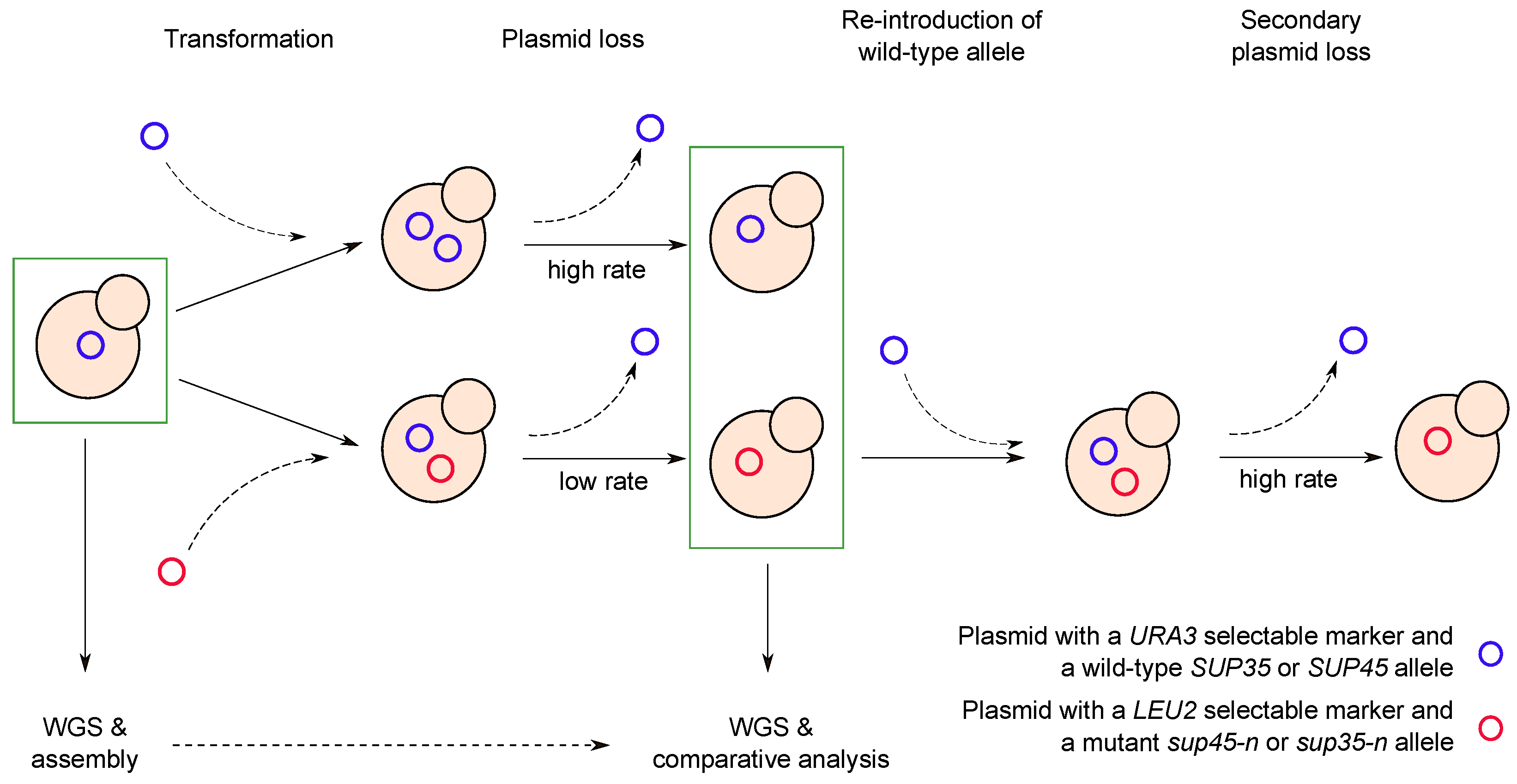
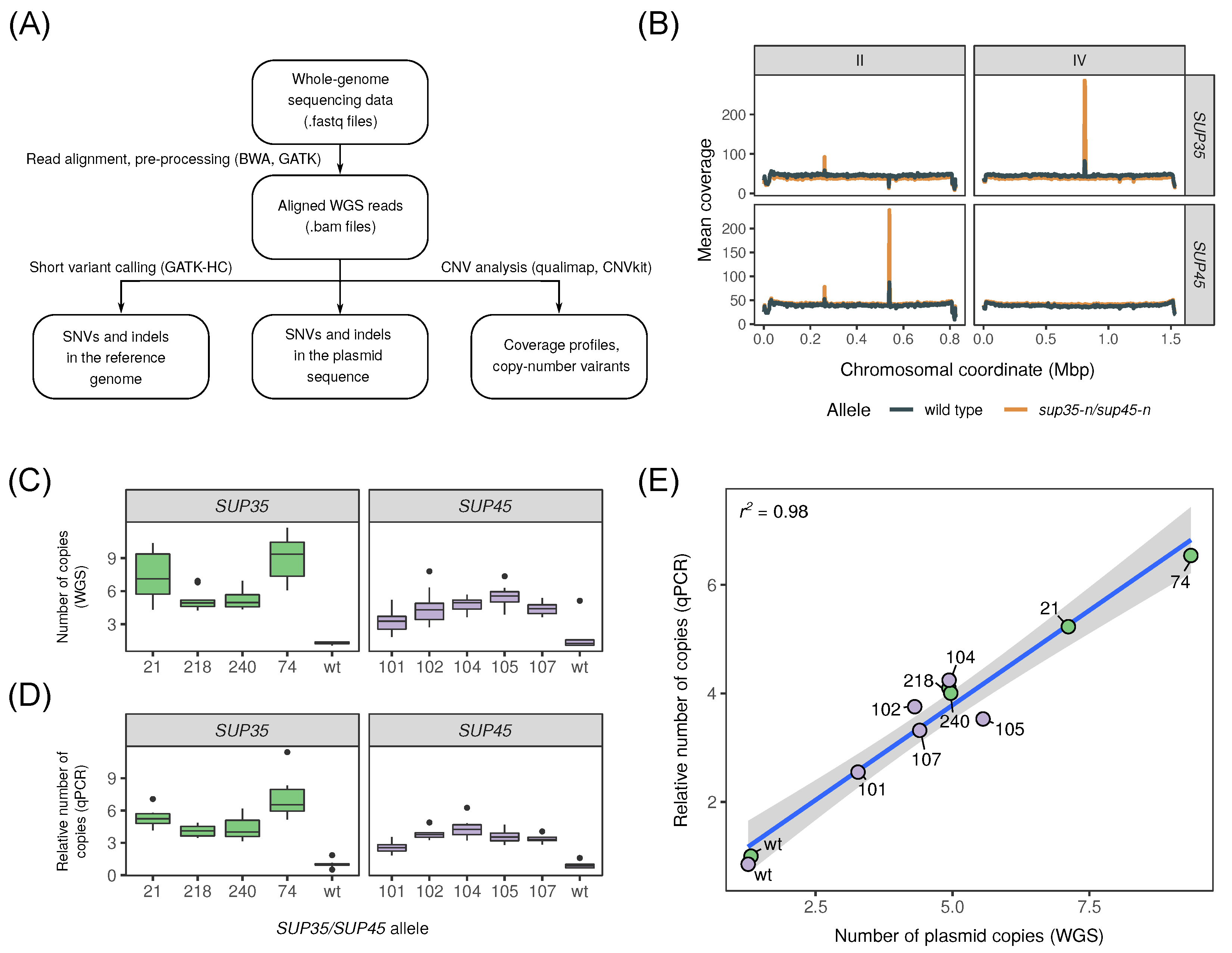
:
1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Media
2.2. Genomic DNA Extraction
2.3. Whole-Genome Sequencing
2.4. Whole-Genome Sequencing Data Analysis
2.5. RNA Extraction and cDNA Generation
2.6. qPCR
2.7. Statistical Analyses and Code Availability
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| PGC | Peterhof Genetic Collection |
| WGS | Whole genome sequencing |
| eRF1/eRF3 | eukaryotic release factor 1/3 |
| 5-FOA | 5-fluoroorotic acid |
References
- Huang, L.; Aghajan, M.; Quesenberry, T.; Low, A.; Murray, S.F.; Monia, B.P.; Guo, S. Targeting translation termination machinery with antisense oligonucleotides for diseases caused by nonsense mutations. Nucleic Acid Ther. 2019, 29, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalder, L.; Mühlemann, O. The meaning of nonsense. Trends Cell Biol. 2008, 18, 315–321. [Google Scholar] [CrossRef]
- Nagel-Wolfrum, K.; Möller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting nonsense mutations in diseases with translational read-through-inducing drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- Liu, J.; Liu, D.; Li, M.; Wu, K.; Liu, N.; Zhao, C.; Shi, X.; Liu, Q. Identification of a nonsense mutation in TNNI3K associated with cardiac conduction disease. J. Clin. Lab. Anal. 2020, 34, 1–7. [Google Scholar] [CrossRef]
- Inge-Vechtomov, S.G.; Mironova, L.N.; Ter-Avanesian, M.D. Ambiguity of translation: An eukaryotic version? Genetika 1994, 30, 1022–1035. [Google Scholar]
- Frolova, L.; Le Goff, X.; Rasmussen, H.H.; Cheperegin, S.; Drugeon, G.; Kress, M.; Arman, I.; Haenni, A.L.; Celis, J.E.; Philippe, M. A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature 1994, 372, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [CrossRef]
- Stansfield, I.; Jones, K.M.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Ter-Avanesyan, M.D.; Telckov, M.V.; Surguchov, A.P.; Smirnov, V.N.; Inge-Vechtomov, S.G. Nucleotide sequence of the SUP2 (SUP35) gene of Saccharomyces cerevisiae. Gene 1988, 66, 45–54. [Google Scholar] [CrossRef]
- Moskalenko, S.E.; Chabelskaya, S.V.; Inge-Vechtomov, S.G.; Philippe, M.; Zhouravleva, G.A. Viable nonsense mutants for the essential gene SUP45 of Saccharomyces cerevisiae. BMC Mol. Biol. 2003, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chabelskaya, S.; Kiktev, D.; Inge-Vechtomov, S.; Philippe, M.; Zhouravleva, G. Nonsense mutations in the essential gene SUP35 of Saccharomyces cerevisiae are non-lethal. Mol. Genet. Genom. MGG 2004, 272, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Derkatch, I.L.; Uptain, S.M.; Patino, M.M.; Lindquist, S.; Liebman, S.W. The yeast non-Mendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J. 1999, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, M.E.; Bagriantsev, S.; Vishveshwara, N.; Liebman, S.W. Guanidine reduces stop codon read-through caused by missense mutations in SUP35 or SUP45. Yeast 2003, 20, 625–632. [Google Scholar] [CrossRef]
- Stansfield, I.; Eurwilaichitr, L.; Akhmaloka.; Tuite, M.F. Depletion in the levels of the release factor eRF1 causes a reduction in the efficiency of translation termination in yeast. Mol. Microbiol. 1996, 20, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Masison, D.C.; Edskes, H.K. [PSI] and [URE3] as yeast prions. Yeast 1995, 11, 1671–1685. [Google Scholar] [CrossRef]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef]
- Trubitsina, N.; Zemlyanko, O.; Moskalenko, S.; Zhouravleva, G. From past to future: Suppressor mutations in yeast genes encoding translation termination factors. Biol. Commun. 2019, 64, 89–109. [Google Scholar] [CrossRef]
- Betney, R.; De Silva, E.; Mertens, C.; Knox, Y.; Krishnan, J.; Stansfield, I. Regulation of release factor expression using a translational negative feedback loop: A systems analysis. RNA 2012, 18, 2320–2334. [Google Scholar] [CrossRef] [Green Version]
- Zhouravleva, G.A.; Moskalenko, S.E.; Chabelskaya, S.; Philippe, M.; Inge-Vechtomov, S.G. Increased tRNA concentration in yeast containing mutant termination translation factors eRF1 and eRF3. Mol. Biol. 2006, 40, 724–730. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; p. 1626. [Google Scholar]
- Kaiser, C.; Michaelis, S.; Mitchell, A. Methods in Yeast Genetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1994; p. 234. [Google Scholar]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Matveenko, A.G.; Matiiv, A.B.; Maksiutenko, E.M.; Moskalenko, S.E.; Drozdova, P.B.; Polev, D.E.; Beliavskaia, A.Y.; Danilov, L.G.; Predeus, A.V.; et al. Chromosome-level genome assembly and structural variant analysis of two laboratory yeast strains from the Peterhof Genetic Collection lineage. G3 Genes Genomes Genet. 2021, 11, jkab029. [Google Scholar] [CrossRef]
- Volkov, K.V.; Aksenova, A.Y.; Soom, M.J.; Osipov, K.V.; Svitin, A.V.; Kurischko, C.; Shkundina, I.S.; Ter-Avanesyan, M.D.; Inge-Vechtomov, S.G.; Mironova, L.N. Novel non-Mendelian determinant involved in the control of translation accuracy in Saccharomyces cerevisiae. Genetics 2002, 160, 25–36. [Google Scholar] [CrossRef]
- Danilov, L.G.; Matveenko, A.G.; Ryzhkova, V.E.; Belousov, M.V.; Poleshchuk, O.I.; Likholetova, D.V.; Sokolov, P.A.; Kasyanenko, N.A.; Kajava, A.V.; Zhouravleva, G.A.; et al. Design of a new [PSI+]-No-More mutation in SUP35 with strong inhibitory effect on the [PSI+] prion propagation. Front. Mol. Neurosci. 2019, 12, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002, 350, 87–96. [Google Scholar]
- Drozdova, P.B.; Tarasov, O.V.; Matveenko, A.G.; Radchenko, E.A.; Sopova, J.V.; Polev, D.E.; Inge-Vechtomov, S.G.; Dobrynin, P.V. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strains of the Peterhof Genetic Collection. PLoS ONE 2016, 11, e0154722. [Google Scholar] [CrossRef] [Green Version]
- Matveenko, A.G.; Drozdova, P.B.; Moskalenko, S.E.; Tarasov, O.V.; Zhouravleva, G.A. Whole genome sequencing data and analyses of the underlying SUP35 transcriptional regulation for a Saccharomyces cerevisiae nonsense suppressor mutant. Data Brief 2019, 23, 103694. [Google Scholar] [CrossRef]
- Le Goff, C.; Zemlyanko, O.; Moskalenko, S.; Berkova, N.; Inge-Vechtomov, S.; Philippe, M.; Zhouravleva, G. Mouse GSPT2, but not GSPT1, can substitute for yeast eRF3 in vivo. Genes Cells 2002, 7, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Strope, P.K.; Skelly, D.A.; Kozmin, S.G.; Mahadevan, G.; Stone, E.A.; Magwene, P.M.; Dietrich, F.S.; McCusker, J.H. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 2015, 125, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.O.; Sherlock, G.; Petrov, D.A. Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 Genes Genomes Genet. 2016, 6, 2421–2434. [Google Scholar] [CrossRef] [Green Version]
- Drozdova, P.; Mironova, L.; Zhouravleva, G. Haploid yeast cells undergo a reversible phenotypic switch associated with chromosome II copy number. BMC Genet. 2016, 17, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Tschumper, G.; Carbon, J. Copy number control by a yeast centromere. Gene 1983, 23, 221–232. [Google Scholar] [CrossRef]
- Bonney, M.E.; Moriya, H.; Amon, A. Aneuploid proliferation defects in yeast are not driven by copy number changes of a few dosage-sensitive genes. Genes Dev. 2015, 29, 898–903. [Google Scholar] [CrossRef] [Green Version]
- Metzger, M.B.; Scales, J.L.; Dunklebarger, M.F.; Weissman, A.M. The ubiquitin ligase (E3) Psh1p is required for proper segregation of both centromeric and two-micron plasmids in Saccharomyces cerevisiae. G3 Genes Genomes Genet. 2017, 7, 3731–3743. [Google Scholar] [CrossRef] [Green Version]
- Hose, J.; Yong, C.M.; Sardi, M.; Wang, Z.; Newton, M.A.; Gasch, A.P. Dosage compensation can buffer copy-number variation in wild yeast. eLife 2015, 4, e05462. [Google Scholar] [CrossRef]
- Torres, E.M.; Springer, M.; Amon, A. No current evidence for widespread dosage compensation in S. cerevisiae. eLife 2016, 5, e10996. [Google Scholar] [CrossRef]
- Gasch, A.P.; Hose, J.; Newton, M.A.; Sardi, M.; Yong, M.; Wang, Z. Further support for aneuploidy tolerance in wild yeast and effects of dosage compensation on gene copy-number evolution. eLife 2016, 5, e14409. [Google Scholar] [CrossRef] [PubMed]
- Zadorsky, S.P.; Sopova, Y.V.; Andreichuk, D.Y.; Startsev, V.A.; Medvedeva, V.P.; Inge-Vechtomov, S.G. Chromosome VIII disomy influences the nonsense suppression efficiency and transition metal tolerance of the yeast Saccharomyces cerevisiae. Yeast 2015, 32, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.G.; Rockmill, B.M.; Blechl, A.E.; Maloney, D.H.; Resnick, M.A.; Fogel, S. The detection of mitotic and meiotic aneuploidy in yeast using a gene dosage selection system. Mol. Gen. Genet. 1988, 215, 10–18. [Google Scholar] [CrossRef]
- Gong, H.; Romanova, N.V.; Allen, K.D.; Chandramowlishwaran, P.; Gokhale, K.; Newnam, G.P.; Mieczkowski, P.; Sherman, M.Y.; Chernoff, Y.O. Polyglutamine toxicity is controlled by prion composition and gene dosage in yeast. PLoS Genet. 2012, 8, e1002634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, H.; Grimm, M. Misreading of termination codons in eukaryotes by natural nonsense suppressor tRNAs. Nucleic Acids Res. 2001, 29, 4767–4782. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, S.; Cornu, D.; Argentini, M.; Namy, O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 10061–10072. [Google Scholar] [CrossRef] [Green Version]
- Kiktev, D.; Moskalenko, S.; Murina, O.; Baudin-Baillieu, A.; Rousset, J.P.; Zhouravleva, G. The paradox of viable sup45 STOP mutations: A necessary equilibrium between translational readthrough, activity and stability of the protein. Mol. Genet. Genom. 2009, 282, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Betney, R.; De Silva, E.; Krishnan, J.; Stansfield, I. Autoregulatory systems controlling translation factor expression: Thermostat-like control of translational accuracy. RNA 2010, 16, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchsenius, A.S.; Tchourikova, A.A.; Inge-Vechtomov, S.G. Recessive mutations in SUP35 and SUP45 genes coding for translation release factors affect chromosome stability in Saccharomyces cerevisiae. Curr. Genet. 2000, 37, 285–291. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
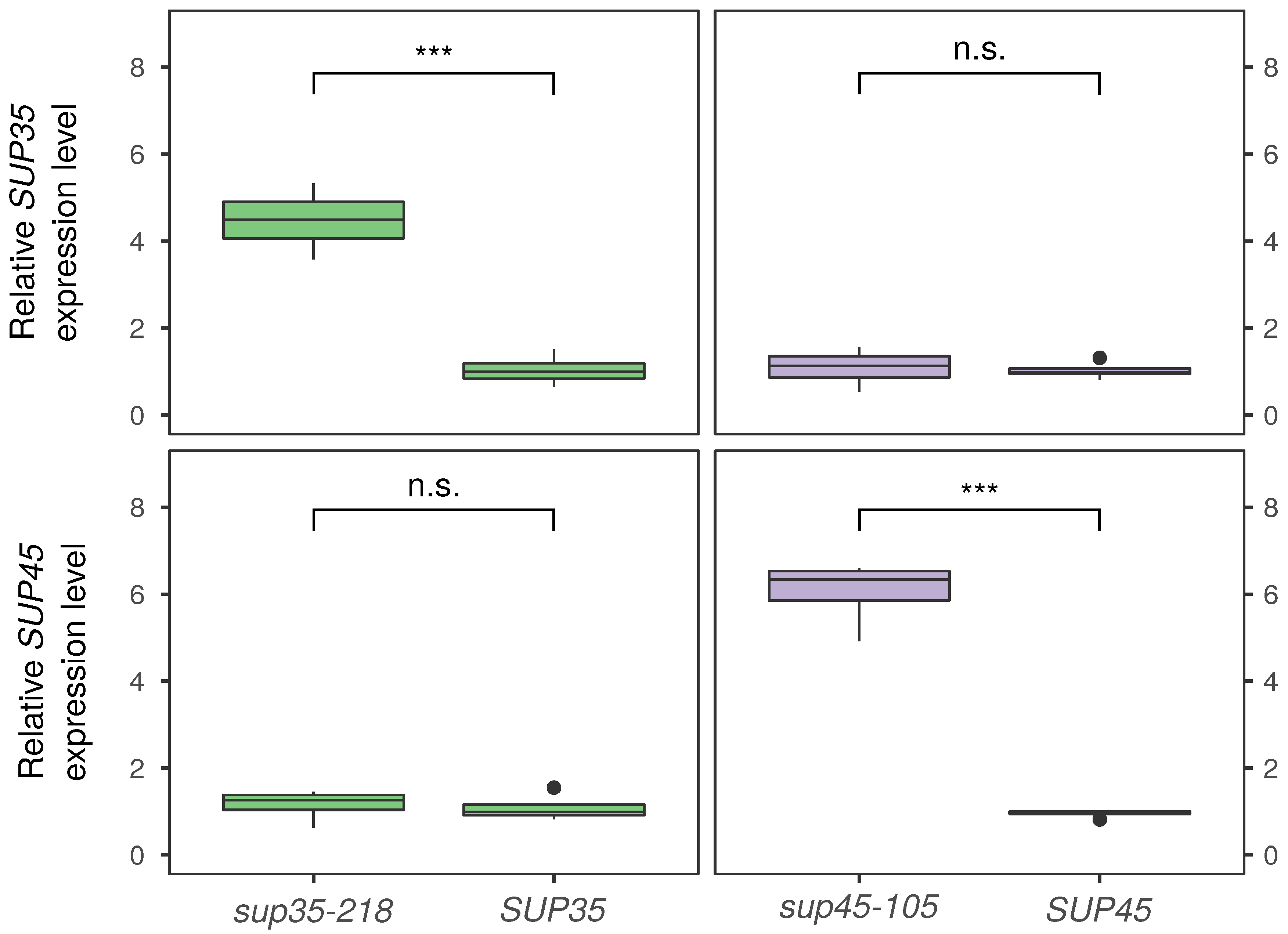{kind=link}
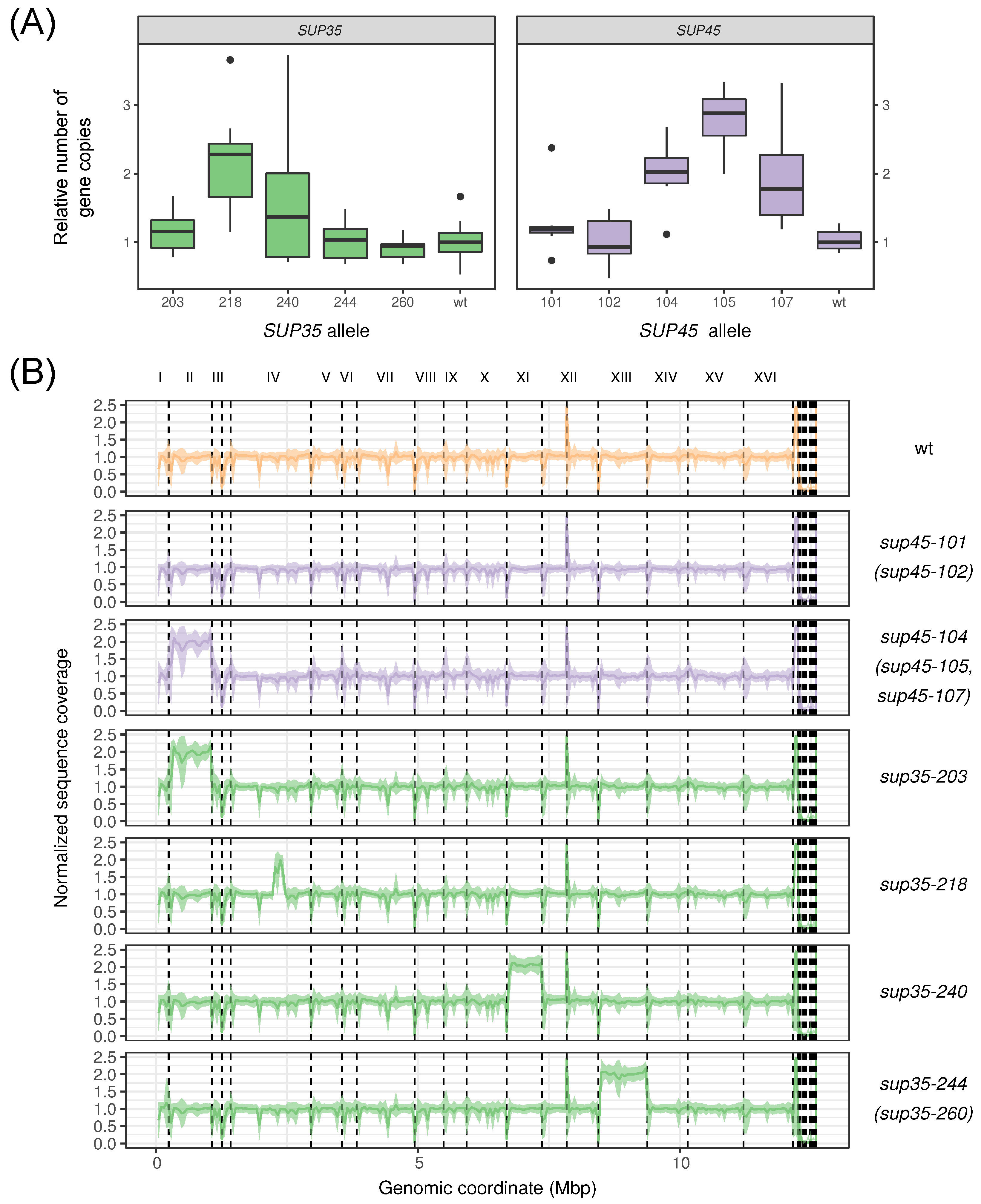{kind=link}
| Strajn | Genotype | References | Sequencing Data |
|---|---|---|---|
| U-1A-D1628 | MATα ade1-14(UGA) trp1-289(UAG) | [11] | [24] |
| his3-Δ200 lys2 ura3-52 leu2-3,112 | |||
| SUP45::HIS3MX [pRS316-SUP45] | |||
| L-1A-D1628 | MATα ade1-14(UGA) trp1-289(UAG) | [11] | This work |
| his3-Δ200 lys2 ura3-52 leu2-3,112 | |||
| SUP45::HIS3MX [pRS315-SUP45] | |||
| nL-1A-D1628 | MATα ade1-14(UGA) trp1-289(UAG) | [11] | This work |
| his3-Δ200 lys2 ura3-52 leu2-3,112 | |||
| SUP45::HIS3MX [pRS315-sup45-n] | |||
| U-14-D1690 | MATα ade1-14(UGA) trp1-289(UAG) | This work | This work |
| his3-Δ200 lys2 ura3-52 leu2-3,112 | |||
| SUP35::HIS3MX [pRSU2] | |||
| L-14-D1690 | MATα ade1-14(UGA) trp1-289(UAG) | This work | This work |
| his3-Δ200 lys2 ura3-52 leu2-3,112 | |||
| SUP35::HIS3MX [pRSU1] | |||
| nL-14-D1690 | MATα ade1-14(UGA) trp1-289(UAG) | This work | This work |
| his3-Δ200 lys2 ura3-52 leu2-3,112 | |||
| SUP35::HIS3MX [pRSU1-n] | |||
| 1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [11] | [28]; This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B | |||
| 101-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [11] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup45-101 | |||
| 102-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [11] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup45-102 | |||
| 104-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [11] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup45-104 | |||
| 105-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [11] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup45-105 | |||
| 107-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [11] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup45-107 | |||
| 222-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [12] | [29] |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup35-222 | |||
| 203-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [12] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup35-203 | |||
| 218-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [12] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup35-218 | |||
| 240-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [12] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup35-240 | |||
| 244-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [12] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup35-244 | |||
| 260-1B-D1606 | MATα ade1-14(UGA) his7-1(UAA) | [12] | This work |
| lys9-A21(UAA) trp1-289(UAG) ura3-52 | |||
| leu2–3,112 gal10-1B sup35-260 |
| Plasmid | Description (Selectable Marker, Promoter, Gene) | References |
|---|---|---|
| pRS316-SUP45 | URA3, PSUP45, SUP45 | [30] |
| pRS315-SUP45 | LEU2, PSUP45, SUP45 | [30] |
| pRS315-sup45-101 | LEU2, PSUP45, sup45-101 (G796T) (E266[UAA]) | [11] |
| pRS315-sup45-102 | LEU2, PSUP45, sup45-102 (T159A) (Y53[UAA]) | [11] |
| pRS315-sup45-104 | LEU2, PSUP45, sup45-104 (T848A) (L283[UAA]) | [11] |
| pRS315-sup45-105 | LEU2, PSUP45, sup45-105 (G1153T) (E385[UAA]) | [11] |
| pRS315-sup45-107 | LEU2, PSUP45, sup45-107 (T950G) (L317[UGA]) | [11] |
| pRSU2 | URA3, PSUP35, SUP35 | [25] |
| pRSU1 | LEU2, PSUP35, SUP35 | [25] |
| pRSU1-21 | LEU2, PSUP35, sup35-21 (C1264T) (Q422[UAA]) | [12] |
| pRSU1-74 | LEU2, PSUP35, sup35-74 (C388E) (Q130[UAA]) | [12] |
| pRSU1-218 | LEU2, PSUP35, sup35-218 (G541T) (E181[UAA]) | [12] |
| pRSU1-240 | LEU2, PSUP35, sup35-240 (C166T) (Q56[UAA]) | [12] |
| No. | Primer Name | Target Gene | Sequences |
|---|---|---|---|
| 1 | bla_F | bla | ATAAATCTGGAGCCGGTGAG |
| 2 | bla_R | bla | CTACGATACGGGAGGGCTTA |
| 3 | SUP45_F | SUP45 | CGATCCAAGACTAGCATGTAAG |
| 4 | SUP45_R | SUP45 | CTTGAACATACTTGACATTGGC |
| 5 | SUP35_F | SUP35 | ACAACAAGGTAACAACAGATACC |
| 6 | SUP35_R | SUP35 | GGATTGAATTGCTGCTGATAAC |
| 7 | ACT1_F | ACT1 | TAACGGTTCTGGTATGTGTAAAGC |
| 8 | ACT1_R | ACT1 | GCTTCATCACCAACGTAGGAGTC |
| 9 | F-ADH1-RT | ADH1 | CAAGTCGTCAAGTCCATCTC |
| 10 | R-ADH1-RT | ADH1 | GTAGACAAGCCGACAACCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksiutenko, E.M.; Barbitoff, Y.A.; Matveenko, A.G.; Moskalenko, S.E.; Zhouravleva, G.A. Gene Amplification as a Mechanism of Yeast Adaptation to Nonsense Mutations in Release Factor Genes. Genes 2021, 12, 2019. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122019
Maksiutenko EM, Barbitoff YA, Matveenko AG, Moskalenko SE, Zhouravleva GA. Gene Amplification as a Mechanism of Yeast Adaptation to Nonsense Mutations in Release Factor Genes. Genes. 2021; 12(12):2019. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122019
Chicago/Turabian StyleMaksiutenko, Evgeniia M., Yury A. Barbitoff, Andrew G. Matveenko, Svetlana E. Moskalenko, and Galina A. Zhouravleva. 2021. "Gene Amplification as a Mechanism of Yeast Adaptation to Nonsense Mutations in Release Factor Genes" Genes 12, no. 12: 2019. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122019