
Genome-Wide Association Study Candidate Genes on Mammary System-Related Teat-Shape Conformation Traits in Chinese Holstein Cattle


 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Phenotype Measurement and DNA Sample Collection
2.3. Phenotype and Genotype Parameters for Analysis
2.4. Genotyping and Quality Control
2.5. Population Structure Analysis
2.6. Genome-Wide Association Analysis
2.7. Candidate Gene Identification
2.8. Gene Pathway Enrichment Analysis
3. Results
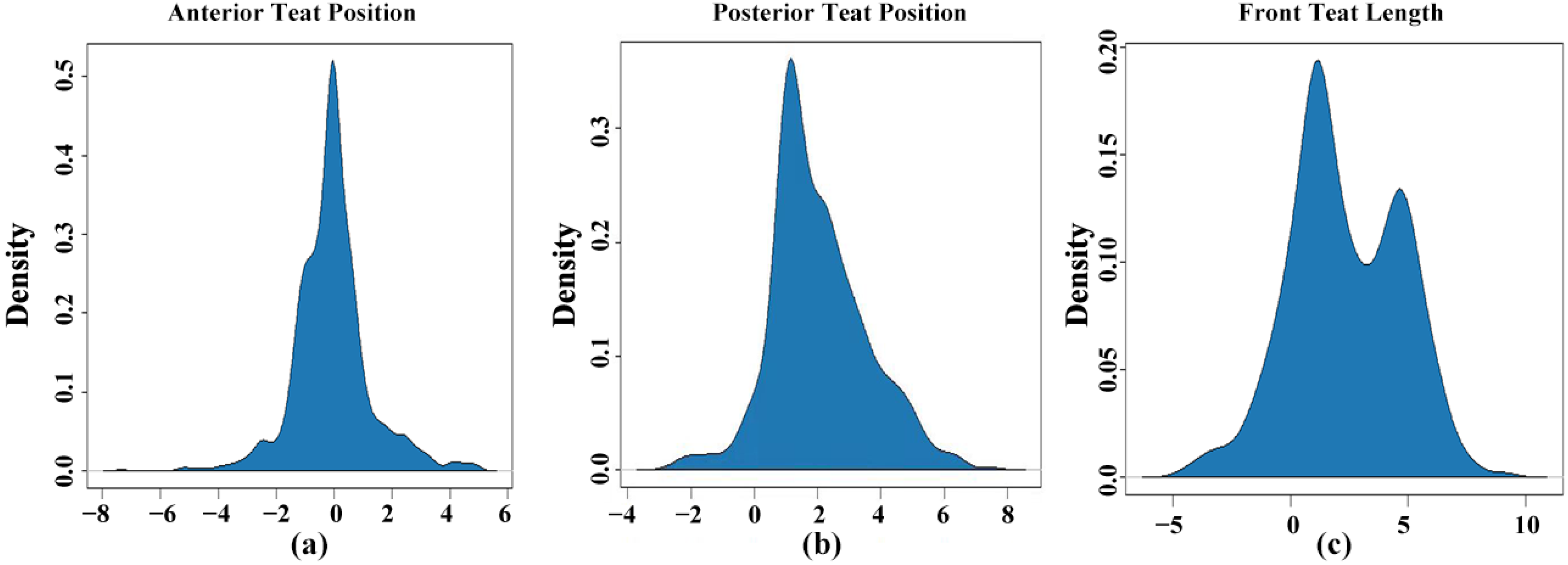
3.1. Description and Heritability of Traits
3.2. Phenotype and Genotype Correlation
3.3. Information of Marker (SNPs)
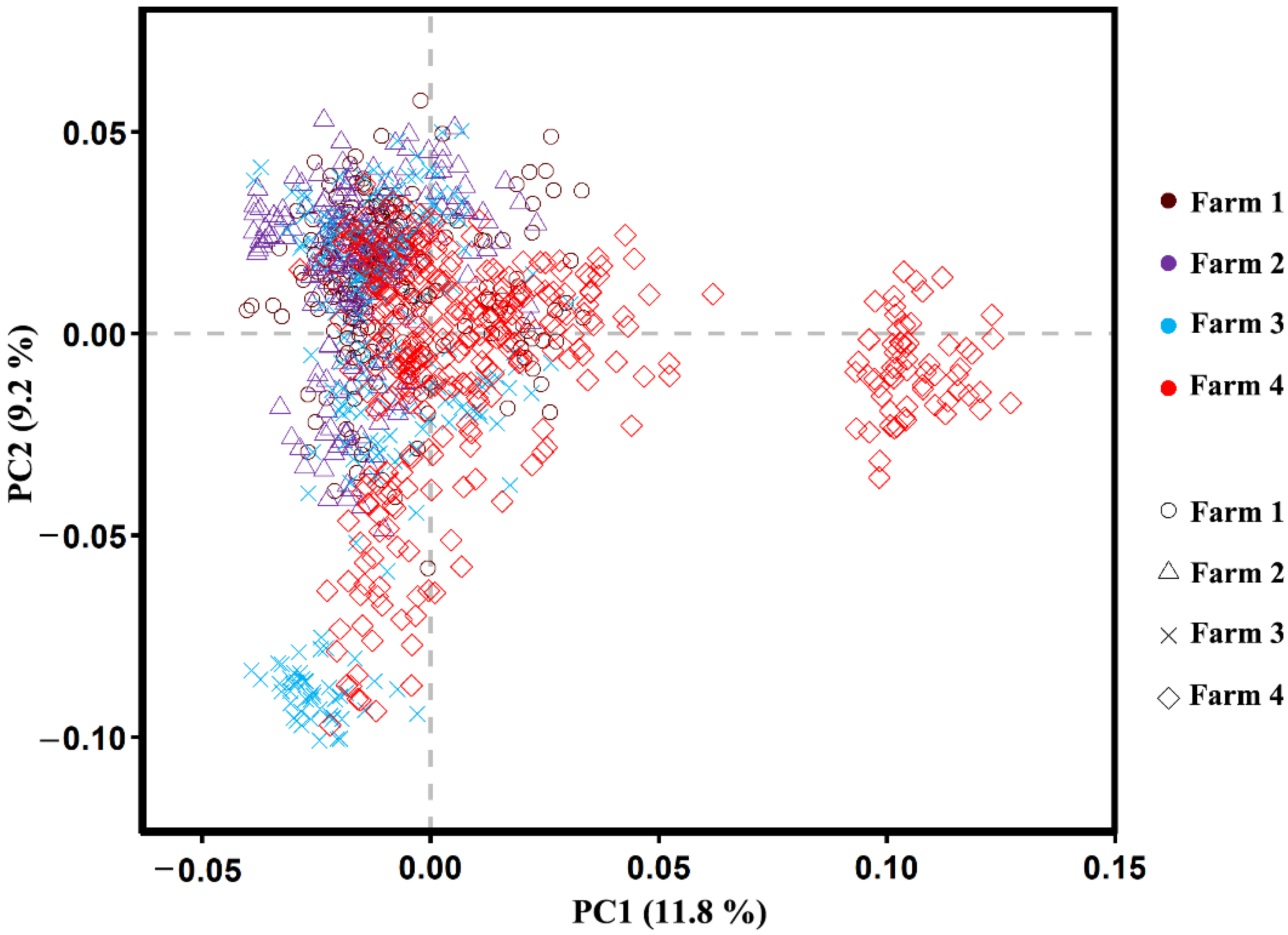
3.4. Population Structure Analysis
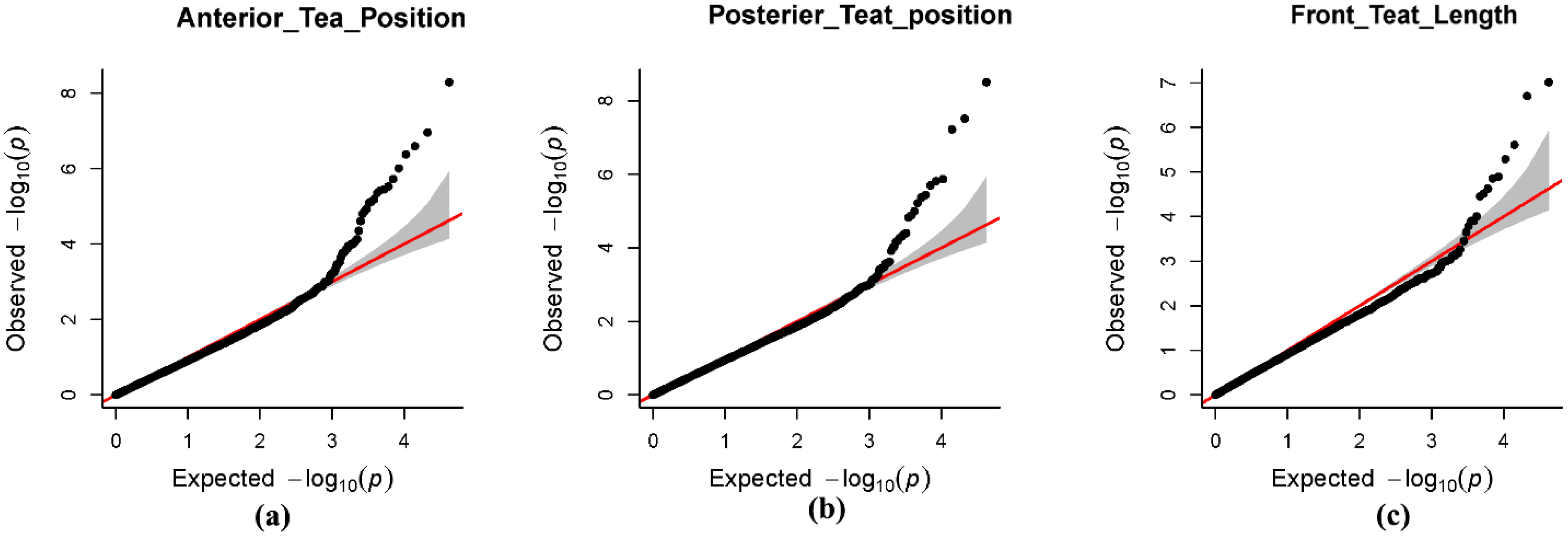
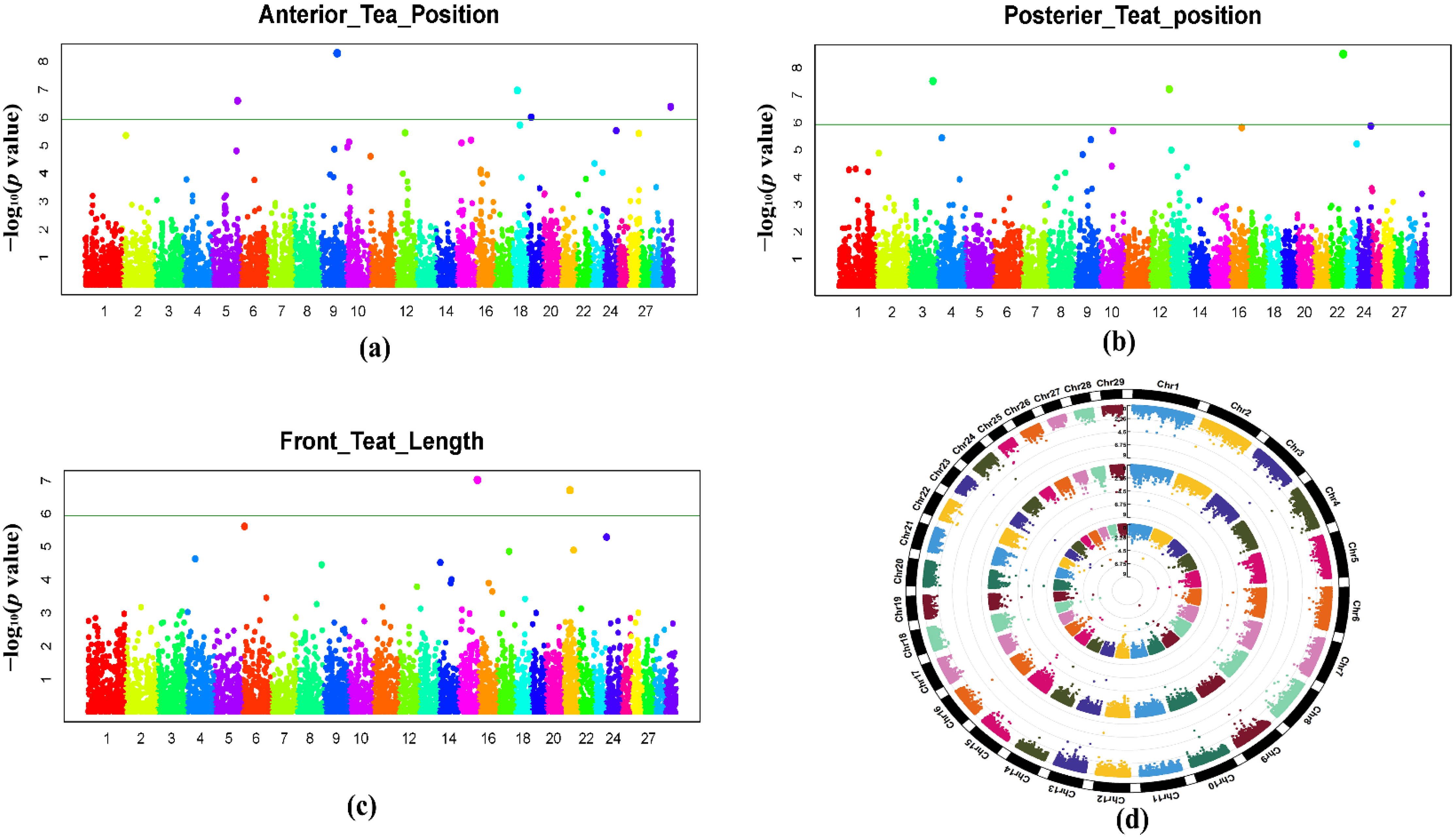
3.5. Results of Genome-Wide Associations
3.6. Identification of Candidate Gene
3.7. Bioinformatics Analysis
3.8. Gene Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Devani, K.; Plastow, G.; Orsel, K.; Valente, T.S. Genome-wide association study for mammary structure in Canadian Angus cows. PLoS ONE 2020, 15, e0237818. [Google Scholar] [CrossRef] [PubMed]
- Tiezzi, F.; Maisano, A.M.; Chessa, S.; Luini, M.; Biffani, S. Heritability of Teat Condition in Italian Holstein Friesian and Its Relationship with Milk Production and Somatic Cell Score. Animals 2020, 10, 2271. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Singh, N.K.; Bhadwal, M.S. Relationship of Somatic Cell Count and Mastitis: An Overview. Asian-Australas. J. Anim. Sci. 2011, 24, 429–438. [Google Scholar] [CrossRef]
- Klein, D.; Flöck, M.; Khol, J.L.; Franz, S.; Stüger, H.P.; Baumgartner, W. Ultrasonographic measurement of the bovine teat: Breed differences, and the significance of the measurements for udder health. J. Dairy Res. 2005, 72, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.; Weinfurtner, M.; Bruckmaier, R. Teat Anatomy and its Relationship with Quarter and Udder Milk Flow Characteristics in Dairy Cows. J. Dairy Sci. 2004, 87, 3280–3289. [Google Scholar] [CrossRef]
- More, S.J.; McKenzie, K.; O’Flaherty, J.; Doherty, M.L.; Cromie, A.R.; Magan, M.J. Setting priorities for non-regulatory animal health in Ireland: Results from an expert Policy Delphi study and a farmer priority identification survey. Prev. Vet. Med. 2010, 95, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, K.; Trajcev, M.; Buneski, G. A review of the factors affecting the costs of bovine mastitis: Review article. J. S. Afr. Vet. Assoc. 2006, 77, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapinal, N.; Liang, Y.; Weary, D.; Wang, Y.; Von Keyserlingk, M. Risk factors for lameness and hock injuries in Holstein herds in China. J. Dairy Sci. 2014, 97, 4309–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, D.; Rogers, G.; Cooper, J.; Hargrove, G.; Keown, J.; Hansen, L. Heritability of Clinical Mastitis Incidence and Relationships with Sire Transmitting Abilities for Somatic Cell Score, Udder Type Traits, Productive Life, and Protein Yield. J. Dairy Sci. 2000, 83, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Lund, T.; Miglior, F.; Dekkers, J.; Burnside, E. Genetic relationships between clinical mastitis, somatic cell count, and udder conformation in Danish Holsteins. Livest. Prod. Sci. 1994, 39, 243–251. [Google Scholar] [CrossRef]
- Abdalla, I.M.; Lu, X.; Nazar, M.; Adam, A.; Arbab, I.; Xu, T.; Yousif, M.H.; Mao, Y.; Yang, Z. Genome-Wide Association Study Identifies Candidate Genes Holstein Cattle. Animals 2021, 11, 2259. [Google Scholar] [CrossRef] [PubMed]
- Chrystal, M.A.; Seykora, A.J.; Hansen, L.B. Heritabilities of Teat End Shape and Teat Diameter and Their Relationships with Somatic Cell Score. J. Dairy Sci. 1999, 82, 2017–2022. [Google Scholar] [CrossRef]
- Visscher, P.; Goddard, M. Genetic Parameters for Milk Yield, Survival, Workability, and Type Traits for Australian Dairy Cattle. J. Dairy Sci. 1995, 78, 205–220. [Google Scholar] [CrossRef]
- Wang, D.; Ning, C.; Liu, J.-F.; Zhang, Q.; Jiang, L. Short communication: Replication of genome-wide association studies for milk production traits in Chinese Holstein by an efficient rotated linear mixed model. J. Dairy Sci. 2019, 102, 2378–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ma, P.; Liu, J.; Zhang, Q.; Zhang, Y.; Ding, X.; Jiang, L.; Wang, Y.; Zhang, Y.; Sun, D.; et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genom. 2011, 12, 408. [Google Scholar] [CrossRef] [Green Version]
- Vollema, A.; Van Der Beek, S.; Harbers, A.; De Jong, G. Genetic Evaluation for Longevity of Dutch Dairy Bulls. J. Dairy Sci. 2000, 83, 2629–2639. [Google Scholar] [CrossRef]
- Silva, A.; Azevedo, A.; Verneque, R.; Gasparini, K.; Peixoto, M.; da Silva, M.; Lopes, P.; Guimarães, S.; Machado, M. Quantitative trait loci affecting milk production traits on bovine chromosome 6 in zebuine Gyr breed. J. Dairy Sci. 2011, 94, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, A.C.C.; Guimarães, S.E.F.; Kelly, M.J.; Fortes, M.R.S.; e Silva, F.F.; Verardo, L.L.; Mota, R.; Moore, S. Multiple-trait genomewide mapping and gene network analysis for scrotal circumference growth curves in Brahman cattle. J. Anim. Sci. 2017, 95, 3331. [Google Scholar] [CrossRef] [PubMed]
- Carlström, C.; Pettersson, G.; Johansson, K.; Strandberg, E.; Stålhammar, H.; Philipsson, J. Feasibility of using automatic milking system data from commercial herds for genetic analysis of milkability. J. Dairy Sci. 2013, 96, 5324–5332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.L.W.J.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimarães, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50, 40. [Google Scholar] [CrossRef] [Green Version]
- Marete, A.; Lund, M.S.; Boichard, D.; Ramayo-Caldas, Y. A system-based analysis of the genetic determinism of udder conformation and health phenotypes across three French dairy cattle breeds. PLoS ONE 2018, 13, e0199931. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.; Jensen, J.A. A User’S Guide to DMU. A Package for Analysing Multivariate Mixed Models. Available online: https://dmu.ghpc.au.dk/dmu/index.html (accessed on 25 July 2021).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.; Price, A.L.; Patterson, N. Principal component analysis of genetic data. Nat. Genet. 2008, 40, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, L.; Wang, W.; Zhang, S.; Yin, Z.; Zhang, Q.; Liu, J.-F. Associations between variants of the HALgene and milk production traits in Chinese Holstein cows. BMC Genet. 2014, 15, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bland, J.M.; Altman, D.G. Statistics notes: Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, J.N.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M.; et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef]
- Carbon, S.; Dietze, H.; Lewis, S.E.; Mungall, C.J.; Munoz-Torres, M.C.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Fey, P.; Thomas, P.D.; et al. Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Bradford, H.L.; Moser, D.W.; Bormann, J.M.; Weaber, R.L. Estimation of genetic parameters for udder traits in Hereford cattle. J. Anim. Sci. 2015, 93, 2663–2668. [Google Scholar] [CrossRef]
- Wu, X.; Fang, M.; Liu, L.; Wang, S.; Liu, J.; Ding, X.; Zhang, S.; Zhang, Q.; Zhang, Y.; Qiao, L.; et al. Genome wide association studies for body conformation traits in the Chinese Holstein cattle population. BMC Genom. 2013, 14, 897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dochtermann, N.A. Testing Cheverud’s conjecture for behavioral correlations and behavioral syndromes. Evol. Int. J. Org. Evol. 2011, 65, 1814–1820. [Google Scholar] [CrossRef]
- Olasege, B.; Zhang, S.; Zhao, Q.; Liu, D.; Sun, H.; Wang, Q.; Ma, P.; Pan, Y. Genetic parameter estimates for body conformation traits using composite index, principal component, and factor analysis. J. Dairy Sci. 2019, 102, 5219–5229. [Google Scholar] [CrossRef]
- Haldar, T.; Ghosh, S. Effect of Population Stratification on False Positive Rates of Population-Based Association Analyses of Quantitative Traits. Ann. Hum. Genet. 2012, 76, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devlin, B.; Roeder, K. Genomic Control for Association Studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lee, J.S.; Dang, C.G.; Sudrajad, P.; Kim, H.C.; Yeon, S.H.; Kang, H.S.; Lee, S.H. Stories and Challenges of Genome Wide Association Studies in Livestock—A Review. Asian-Australas. J. Anim. Sci. 2015, 28, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef]
- Marai, I.; Haeeb, A. Buffalo’s biological functions as affected by heat stress—A review. Livest. Sci. 2010, 127, 89–109. [Google Scholar] [CrossRef]
- Wang, D.; Sun, Y.; Stang, P.; Berlin, J.A.; Wilcox, M.A.; Li, Q. Comparison of methods for correcting population stratification in a genome-wide association study of rheumatoid arthritis: Principal-component analysis versus multidimensional scaling. BMC Proc. 2009, 3, S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L-TONSL Complex that Promotes Homologous Recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-H.; Ueda, K.; Nakamura, Y.; Daigo, Y. Identification of a novel oncogene, MMS22L, involved in lung and esophageal carcinogenesis. Int. J. Oncol. 2012, 41, 1285–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutik, S.; Rissler, M.; Guan, X.L.; Guiard, B.; Shui, G.; Gebert, N.; Heacock, P.N.; Rehling, P.; Dowhan, W.; Wenk, M.R.; et al. The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. J. Cell Biol. 2008, 183, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, N.; Tamura, S.; Furuki, S.; Miyata, N.; Moser, A.; Shimozawa, N.; Moser, H.W.; Suzuki, Y.; Kondo, N.; Fujiki, Y. Mutations in Novel Peroxin Gene PEX26 That Cause Peroxisome-Biogenesis Disorders of Complementation Group 8 Provide a Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2003, 73, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Venturini, G.C.; Cardoso, D.F.; Baldi, F.; Freitas, A.C.; Aspilcueta-Borquis, R.R.; Santos, D.J.; de Camargo, G.M.F.; Stafuzza, N.B.; Albuquerque, L.G.; Tonhati, H. Association between single-nucleotide polymorphisms and milk production traits in buffalo. Genet. Mol. Res. 2014, 13, 10256–10268. [Google Scholar] [CrossRef]
- Gonzalez, M.; Villa, R.; Villa, C.; Gonzalez, V.; Montano, M.; Medina, G.; Mahadevan, P. Inspection of real and imputed genotypes reveled 76 SNPs associated to rear udder height in Holstein cattle. J. Adv. Vet. Anim. Res. 2020, 7, 234–241. [Google Scholar] [CrossRef]
- Abuzenadah, A.M.; Zaher, G.F.; Dallol, A.; Damanhouri, G.A.; Chaudhary, A.G.; Al-Sayes, F.; Gari, M.A.; Alzahrani, M.; Hindawi, S.; Al-Qahtani, M.H. Identification of a novel SBF2 missense mutation associated with a rare case of thrombocytopenia using whole-exome sequencing. J. Thromb. Thrombolysis 2013, 36, 501–506. [Google Scholar] [CrossRef]
- Hicar, M.D.; Liu, Y.; Allen, C.E.; Wu, L.-C. Structure of the Human Zinc Finger Protein HIVEP3: Molecular Cloning, Expression, Exon–Intron Structure, and Comparison with Paralogous Genes HIVEP1 and HIVEP2. Genomics 2001, 71, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.-D.; Qin, G.-Q.; He, H.-C.; Han, Z.-D.; Liang, Y.-X.; Yang, S.-B.; Huang, Y.-Q.; Zhou, L.; Fu, H.; Jiang, F.-N.; et al. Combined overexpression of HIVEP3 and SOX9 predicts unfavorable biochemical recurrence- free survival in patients with prostate cancer. OncoTargets Ther. 2014, 7, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Jiao, B.; Ge, W.; Zhang, X.; Wang, S.; Zhao, H.; Wang, X. Transcriptome sequencing to detect the potential role of long non-coding RNAs in bovine mammary gland during the dry and lactation period. BMC Genom. 2018, 19, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bakhtiarizadeh, M.R.; Mirzaei, S.; Norouzi, M.; Sheybani, N.; Sadi, M.S.V. Identification of Gene Modules and Hub Genes Involved in Mastitis Development Using a Systems Biology Approach. Front. Genet. 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Kondratiuk, I.; Jakhanwal, S.; Jin, J.; Sathyanarayanan, U.; Kroppen, B.; Pobbati, A.V.; Krisko, A.; Ashery, U.; Meinecke, M.; Jahn, R.; et al. PI(4,5)P2-dependent regulation of exocytosis by amisyn, the vertebrate-specific competitor of synaptobrevin 2. Proc. Natl. Acad. Sci. USA 2020, 117, 13468–13479. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005, 33, 5458–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, B.C.; Zhang, L.; Schütz, L.F.; Perego, M.C.; Maylem, E.R.S.; Spicer, L.J. Regulation of the transcription factor E2F8 gene expression in bovine ovarian cells. Mol. Cell. Endocrinol. 2019, 498, 110572. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zhang, H.-L.; Li, Z.-X.; Liu, Y.; Liu, X.-L. Expression, SNV identification, linkage disequilibrium, and combined genotype association analysis of the muscle-specific gene CSRP3 in Chinese cattle. Gene 2014, 535, 17–23. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, C.; Wang, G.; Ji, Z.; Liu, Z.; Chao, T.; Zhang, S.; Wang, J. Molecular cloning, characterization and tissue specificity of the expression of the ovine CSRP2 and CSRP3 genes from Small-tail Han sheep (Ovis aries). Gene 2016, 580, 47–57. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, X.; Zhou, G.; Barnes, E.L.; Luo, G. Recql5 Plays an Important Role in DNA Replication and Cell Survival after Camptothecin Treatment. Mol. Biol. Cell 2009, 20, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Jiang, M. Metabolomic profiles in yak mammary gland tissue during the lactation cycle. PLoS ONE 2019, 14, e0219220. [Google Scholar] [CrossRef] [PubMed]
- Moro, J.; Tome, D.; Schmidely, P.; Demersay, T.; Azzout-Marniche, D. Histidine: A systematic review on metabolism and physiological effects in human and different animal species. Nutrients 2020, 12, 1414. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, L.; Gao, Y.; Shi, L.; Li, Y.; Liang, W.; Sun, D. Determination of genetic associations between indels in 11 candidate genes and milk composition traits in Chinese Holstein population. BMC Genet. 2019, 20, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, M.M.; Ettenberg, S.A.; Lowrey, G.A.; Russell, E.K.; Lipkowitz, S. Fas expression and function in normal and malignant breast cell lines. Cancer Res. 1996, 56, 4791–4798. [Google Scholar] [PubMed]
- Yue, S.; Wang, Z.; Wang, L.; Peng, Q.; Xue, B. Transcriptome Functional Analysis of Mammary Gland of Cows in Heat Stress and Thermoneutral Condition. Animals 2020, 10, 1015. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Mean | Std. Error | Std. Deviation | Minimum | Maximum | Heritability |
|---|---|---|---|---|---|---|
| ATP | 5.34 | 0.33 | 1.04 | 1 | 9 | 0.17 |
| PTP | 6.50 | 0.51 | 1.62 | 1 | 9 | 0.37 |
| FTL | 5.72 | 0.52 | 1.63 | 2 | 9 | 0.13 |
| Traits | ATP | PTP | FTL |
|---|---|---|---|
| ATP | 1 | 0.27 ** | 0.16 ** |
| PTP | −0.09 | 1 | 0.03 |
| FTL | 0.48 | 0.24 | 1 |
| Traits | SNP Name | CHR | Position (bp) | MAF | Nearest Gene | Distance (kb) | p Value | Effect |
|---|---|---|---|---|---|---|---|---|
| ATP | ARS-BFGL-NGS-101241 | 9 | 67971873 | 0.24 | MMS22L | 100kb | 5.10 × 10−9 | −0.22498 |
| ARS-BFGL-NGS-43147 | 29 | 37332830 | 0.12 | E2F8 | 100kb | 4.16 × 10−7 | −0.24097 | |
| BovineHD1800006781 | 18 | 22013861 | 0.48 | CDH11 | 100kb | 1.09 × 10−7 | 0.16219 | |
| BovineHD0500031672 | 5 | 109356610 | 0.18 | PEX26 | Within | 2.54 × 10−7 | −0.20494 | |
| PTP | ARS-BFGL-NGS-16048 | 22 | 55808310 | 0.34 | TAMM41 | Within | 3.14 × 10−9 | −0.50077 |
| Hapmap58721-rs29026738 | 3 | 104278203 | 0.084 | HIVEP3 | Within | 3.05 × 10−8 | −0.83939 | |
| 12-88054488-G-A-rs42352402 | 12 | 84040288 | 0.168029 | MYO16 | Within | 6.02 × 10−8 | 0.61091 | |
| FTL | BovineHD1500023818 | 15 | 80481458 | 0.37 | SBF2 | Within | 9.69 × 10−8 | 0.27188 |
| BovineHD2100009187 | 21 | 31317530 | 0.46 | STXBP6 | 100kb | 1.98 × 10−7 | 0.28855 |
| KEGG ID | Description | Gene Count | p Value | Genes Name |
|---|---|---|---|---|
| bta05330 | Allograft rejection | 3 | 3.68 × 10−6 | FASLG, BOLA-DRA GZMB |
| bta04210 | Apoptosis | 7 | 4.50 × 10−4 | BID, FASLG, LOC505326, LOC786826, LOC508858, LOC508646, GZMB |
| bta04060 | Cytokine–cytokine receptor interaction | 8 | 1.09 × 10−2 | CCL16, LOC616364, LOC100297044, LOC525415, TNFSF18, FASLG, TNFSF4, TNFSF8 |
| bta00340 | Histidine metabolism | 2 | 4.91 × 10−2 | AMDHD1, HAL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazar, M.; Lu, X.; Abdalla, I.M.; Ullah, N.; Fan, Y.; Chen, Z.; Arbab, A.A.I.; Mao, Y.; Yang, Z. Genome-Wide Association Study Candidate Genes on Mammary System-Related Teat-Shape Conformation Traits in Chinese Holstein Cattle. Genes 2021, 12, 2020. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122020
Nazar M, Lu X, Abdalla IM, Ullah N, Fan Y, Chen Z, Arbab AAI, Mao Y, Yang Z. Genome-Wide Association Study Candidate Genes on Mammary System-Related Teat-Shape Conformation Traits in Chinese Holstein Cattle. Genes. 2021; 12(12):2020. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122020
Chicago/Turabian StyleNazar, Mudasir, Xubin Lu, Ismail Mohamed Abdalla, Numan Ullah, Yongliang Fan, Zhi Chen, Abdelaziz Adam Idriss Arbab, Yongjiang Mao, and Zhangping Yang. 2021. "Genome-Wide Association Study Candidate Genes on Mammary System-Related Teat-Shape Conformation Traits in Chinese Holstein Cattle" Genes 12, no. 12: 2020. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12122020