
Whole Genome Sequencing in the Evaluation of Fetal Structural Anomalies: A Parallel Test with Chromosomal Microarray Plus Whole Exome Sequencing

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
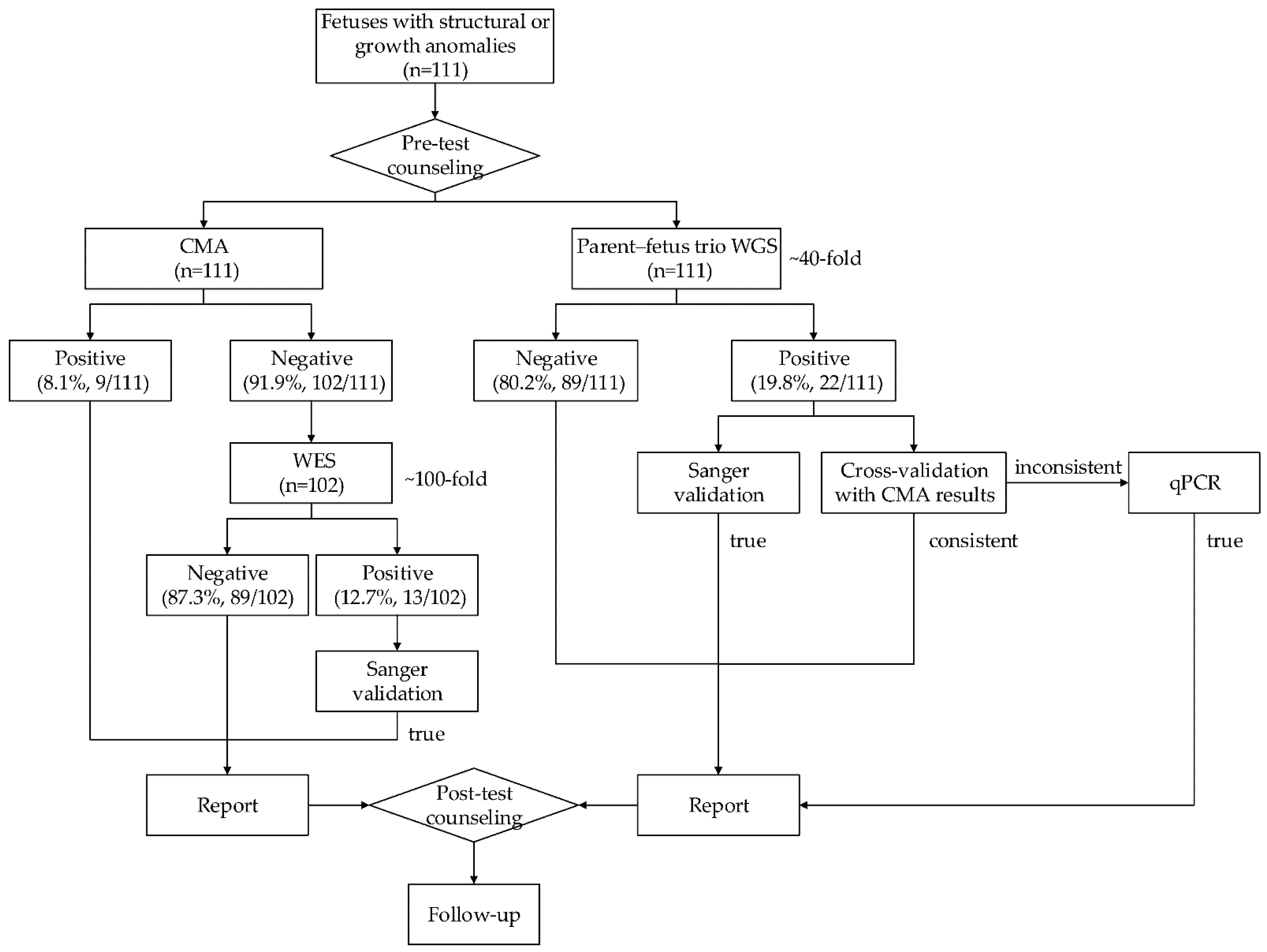
2.1. Study Design and Procedure
2.2. WGS
2.3. CMA
2.4. WES
2.5. Data Interpretation and Reporting
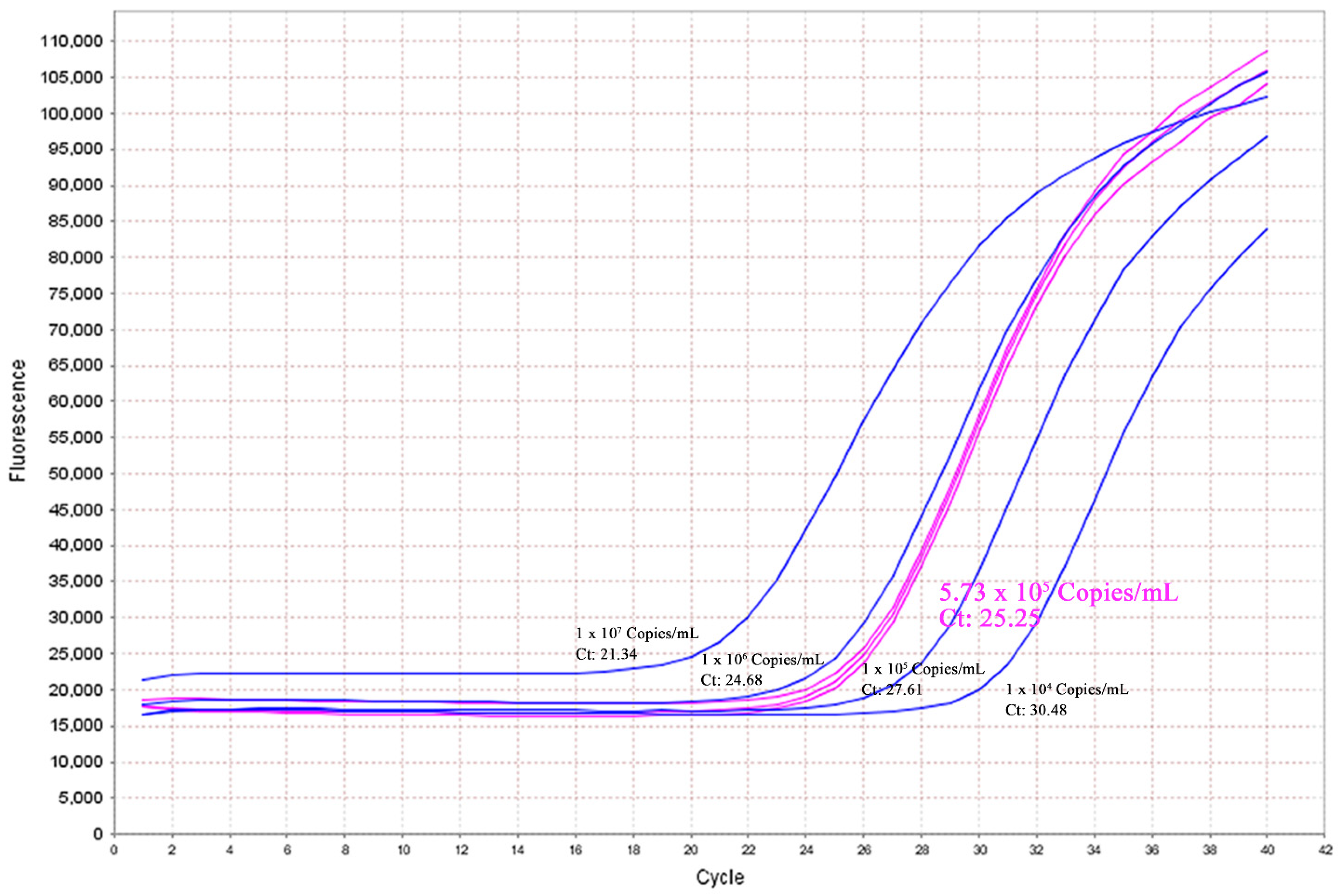
2.6. Data Validation
3. Results
3.1. Study Participants
3.2. Comparison of Clinically Relevant Information Provided by WGS and CMA Plus WES
3.3. Comparison of the CNV/SNV/INDEL Detection by WGS and CMA Plus WES
3.4. Subgroup Analysis
3.5. Impact on Pregnancy Outcome
3.6. Turnaround Time and DNA Requirement
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Approval
References
- Murphy, S.L.; Mathews, T.J.; Martin, J.A.; Minkovitz, C.S.; Strobino, D.M. Annual Summary of Vital Statistics: 2013-2014. Pediatrics 2017, 139. [Google Scholar] [CrossRef] [Green Version]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, J.L.; Shaffer, L.G.; Chitty, L.S.; Rosenfeld, J.A.; Crolla, J.A. The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: A review of the literature. Prenat. Diagn. 2013, 33, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Meier, N.; Bruder, E.; Lapaire, O.; Hoesli, I.; Kang, A.; Hench, J.; Hoeller, S.; De Geyter, J.; Miny, P.; Heinimann, K.; et al. Exome sequencing of fetal anomaly syndromes: Novel phenotype-genotype discoveries. Eur. J. Hum. Genet. 2019, 27, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vora, N.L.; Powell, B.; Brandt, A.; Strande, N.; Hardisty, E.; Gilmore, K.; Foreman, A.K.M.; Wilhelmsen, K.; Bizon, C.; Reilly, J.; et al. Prenatal exome sequencing in anomalous fetuses: New opportunities and challenges. Genet Med. 2017, 19, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Drury, S.; Williams, H.; Trump, N.; Boustred, C.; Gosgene; Lench, N.; Scott, R.H.; Chitty, L.S. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat. Diagn. 2015, 35, 1010–1017. [Google Scholar] [CrossRef] [Green Version]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; McMullan, D.J.; Eberhardt, R.Y.; Rinck, G.; Hamilton, S.J.; Quinlan-Jones, E.; Prigmore, E.; Keelagher, R.; Best, S.K.; Carey, G.K.; et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Aggarwal, V.; Giordano, J.L.; Stosic, M.; Wou, K.; Bier, L.; Spiegel, E.; Brennan, K.; Stong, N.; Jobanputra, V.; et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 2019, 393, 758–767. [Google Scholar] [CrossRef]
- Dharmadhikari, A.V.; Ghosh, R.; Yuan, B.; Liu, P.; Dai, H.; Al Masri, S.; Scull, J.; Posey, J.E.; Jiang, A.H.; He, W.; et al. Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Med. 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, F.; Li, R.; Li, Y.; Nie, Z.Q.; Lei, T.; Wang, D.; Yang, X.; Han, J.; Pan, M.; Zhen, L.; et al. Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obstet. Gynecol. 2018, 51, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Saunders, C.J.; Miller, N.A.; Soden, S.E.; Dinwiddie, D.L.; Noll, A.; Alnadi, N.A.; Andraws, N.; Patterson, M.L.; Krivohlavek, L.A.; Fellis, J.; et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012, 4, 154ra135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willig, L.K.; Petrikin, J.E.; Smith, L.D.; Saunders, C.J.; Thiffault, I.; Miller, N.A.; Soden, S.E.; Cakici, J.A.; Herd, S.M.; Twist, G.; et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: A retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 2015, 3, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Talkowski, M.E.; Ordulu, Z.; Pillalamarri, V.; Benson, C.B.; Blumenthal, I.; Connolly, S.; Hanscom, C.; Hussain, N.; Pereira, S.; Picker, J.; et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N. Engl. J. Med. 2012, 367, 2226–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, K.W.; Wang, H.; Shi, M.; Chen, J.; Yang, Z.; Zhang, R.; Yan, H.; Wang, Y.; Chen, S.; Chau, M.H.K.; et al. Prenatal Diagnosis of Fetuses With Increased Nuchal Translucency by Genome Sequencing Analysis. Front. Genet. 2019, 10, 761. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liang, X.; Xuan, Y.; Geng, C.; Li, Y.; Lu, H.; Qu, S.; Mei, X.; Chen, H.; Yu, T.; et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 2017, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.B.; Xie, Y.J.; Chen, Z.; Zhou, Y.; Wu, J.Z.; Zhang, Z.Q.; Shi, S.S.; Chen, B.J.; Fang, Q. Improved assay performance of single nucleotide polymorphism array over conventional karyotyping in analyzing products of conception. J. Chin. Med. Assoc. 2015, 78, 408–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergant, G.; Maver, A.; Lovrecic, L.; Cuturilo, G.; Hodzic, A.; Peterlin, B. Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: A retrospective survey in 1059 cases. Genet. Med. 2018, 20, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen, SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2019, 22, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, K.G.; Leach, N.T.; Pekarek, D.; Prasad, P.; Rose, N.C.; Practice, A.P.; Guidelines, C. The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of Medical Genetics and Genomics (ACMG). 2020, 10. Genet. Med. 2020, 22, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Barber, J.C.; Rosenfeld, J.A.; Foulds, N.; Laird, S.; Bateman, M.S.; Thomas, N.S.; Baker, S.; Maloney, V.K.; Anilkumar, A.; Smith, W.E.; et al. 8p23.1 duplication syndrome; common, confirmed, and novel features in six further patients. Am. J. Med. Genet. A 2013, 161, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Prokudin, I.; Simons, C.; Grigg, J.R.; Storen, R.; Kumar, V.; Phua, Z.Y.; Smith, J.; Flaherty, M.; Davila, S.; Jamieson, R.V. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1. Eur. J. Hum. Genet. 2014, 22, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, R.R.; Vijayalakshmi, P. Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea. Mol. Vis. 2006, 12, 190–195. [Google Scholar] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [Green Version]
- Chau, M.H.K.; Cao, Y.; Kwok, Y.K.Y.; Chan, S.; Chan, Y.M.; Wang, H.; Yang, Z.; Wong, H.K.; Leung, T.Y.; Choy, K.W. Characteristics and mode of inheritance of pathogenic copy number variants in prenatal diagnosis. Am. J. Obstet. Gynecol. 2019, 221, 493.e1–493.e11. [Google Scholar] [CrossRef]
- Xiang, J.; Ding, Y.; Song, X.; Mao, J.; Liu, M.; Liu, Y.; Huang, C.; Zhang, Q.; Wang, T. Clinical Utility of SNP Array Analysis in Prenatal Diagnosis: A Cohort Study of 5000 Pregnancies. Front Genet. 2020, 11, 571219. [Google Scholar] [CrossRef]
- Corsten-Janssen, N.; Bouman, K.; Diphoorn, J.C.D.; Scheper, A.J.; Kinds, R.; El Mecky, J.; Breet, H.; Verheij, J.; Suijkerbuijk, R.; Duin, L.K.; et al. A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound. Prenat. Diagn. 2020, 40, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Eisfeldt, J.; Pettersson, M.; Vezzi, F.; Wincent, J.; Kaller, M.; Gruselius, J.; Nilsson, D.; Syk Lundberg, E.; Carvalho, C.M.B.; Lindstrand, A. Comprehensive structural variation genome map of individuals carrying complex chromosomal rearrangements. PLoS Genet. 2019, 15, e1007858. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, Z.; He, R.; Qi, J.; Zhang, Z.; Cui, B. Complete Paternal Uniparental Disomy of Chromosome 2 in an Asian Female Identified by Short Tandem Repeats and Whole Genome Sequencing. Cytogenet. Genome Res. 2019, 157, 197–202. [Google Scholar] [CrossRef]
- Lindstrand, A.; Eisfeldt, J.; Pettersson, M.; Carvalho, C.M.B.; Kvarnung, M.; Grigelioniene, G.; Anderlid, B.M.; Bjerin, O.; Gustavsson, P.; Hammarsjo, A.; et al. From cytogenetics to cytogenomics: Whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. 2019, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roach, J.C.; Glusman, G.; Smit, A.F.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weedon, M.N.; Cebola, I.; Patch, A.M.; Flanagan, S.E.; De Franco, E.; Caswell, R.; Rodriguez-Segui, S.A.; Shaw-Smith, C.; Cho, C.H.; Allen, H.L.; et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet. 2014, 46, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Radcliff, J.; Coe, S.J.; Mahon, L.W. Effects of platforms, size filter cutoffs, and targeted regions of cytogenomic microarray on detection of copy number variants and uniparental disomy in prenatal diagnosis: Results from 5026 pregnancies. Prenat. Diagn. 2019, 39, 137–156. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID | Clinical Indication | Serum Screening | NIPS | CMA | WGS | Added Information by WGS |
|---|---|---|---|---|---|---|
| 2 | Cystic hygroma; severe tetralogy of fallot | N | N | arr(21) × 3 | seq(21) × 3 | NA |
| 40 | ventricular septal defect; pulmonary atresia | N | N | arr(21) × 3 | seq(21) × 3 | NA |
| 42 | Ventricular septal defect | N | N | arr(13) × 3 | seq(13) × 3 | NA |
| 51 | Ventricular septal defect; choledochol cyst; ventriculomegaly; echogenic crystalline lens | N | Low-risk | arr[GRCh37]6q25.3q27(158678581-170914297) × 3 | seq[GRCh37]dup(6)(q25.3q27)dn chr6:g.158661896-170927571dup | A pathogenic SNV (NM_005267.4,c.593G>A,p.Arg198Gln) in GJA8 was identified by WGS |
| arr[GRCh37] 8p23.3(158048-2193914) × 1 | seq[GRCh37]del(8)(p23.3)dn chr8:g.156079-2185433del | |||||
| arr[GRCh37]8p23.2p22(2347604-14015208) × 3 | seq[GRCh37]dup(8)(p23.2p22)dn chr8:g.2340301-13996893dup | |||||
| 52 | Congenital heart disease; clubfeet; atrial septal defects; tetralogy of fallot | N | N | arr(21) × 3 | seq(21) × 3 | NA |
| 84 | Holoprosencephaly; ventricular septal defect; polydactyly | N | N | arr(13) × 3 | seq(13) × 3 | NA |
| 93 | single umbilical artery; omphalocele; umbilical cord cyst; clubhands; facial abnormality | N | N | arr(18) × 3 | seq(18) × 3 | NA |
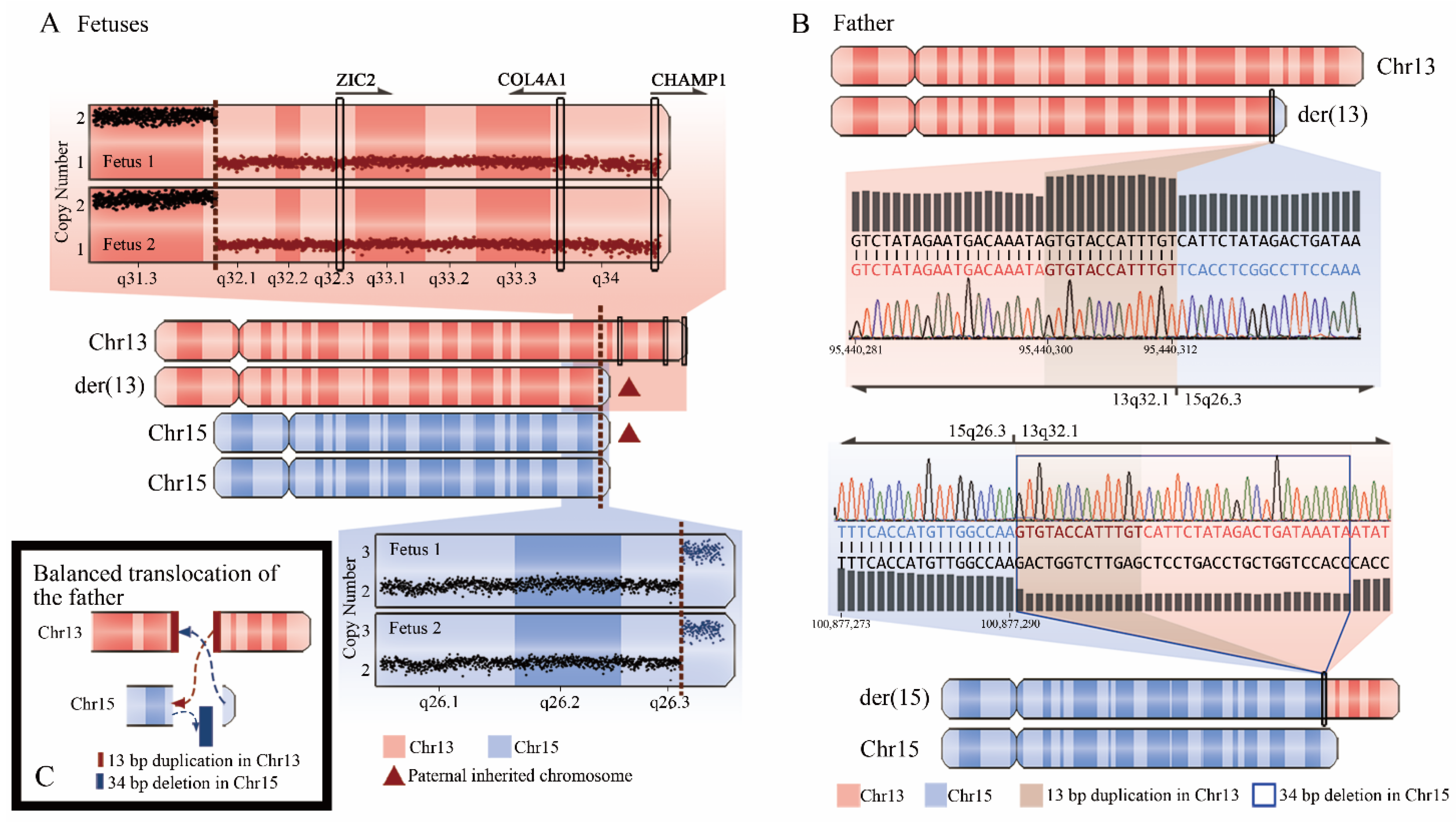
| 102-1 | Skeletal dysplasia; unilateral pleural effusion; intestinal dilatation | N | Low-risk | arr[GRCh37]13q32.1q34(95443088-115107733) × 1 | seq[GRCh37]del(13)(q32.1q34) chr13:g.95440312-115169878del | Arising from paternal balanced translocation |
| arr[GRCh37]15q26.3(100865196-102429040) × 3 | seq[GRCh37]dup(15)(q26.3) chr15:g.100877325-102531392dup | Arising from paternal balanced translocation | ||||
| 102-2 | Fetal hydrops; ascites; pleural effusion; hydroderma | N | Low-risk | arr[GRCh37]13q32.1q34(95443088-115107733) × 1 | seq[GRCh37]del(13)(q32.1q34) chr13:g.95440312-115169878del | Arising from paternal balanced translocation |
| arr[GRCh37]15q26.3(100865196-102429040) × 3 | seq[GRCh37]dup(15)(q26.3) chr15:g.100877325-102531392dup | Arising from paternal balanced translocation |
| Case ID | Clinical Indication | Genes | OMIM Diseases | Variants (All Heterozygous)/Inheritance | Novel or Previously Reported (PMID) |
|---|---|---|---|---|---|
| 1 | Oligohydramnios; renal cyst | PKHD1 | #263200 Polycystic kidney disease 4, with or without hepatic disease (AR) | NM_138694.3, c.7994T>C, p.Leu2665Pro (Mat) | Reported (PMID30507656;PMID28851938) |
| NM_138694.3, c.5428G>T, p.Glu1810* (Pat) | Novel | ||||
| 3 | Skeletal dysplasia; micromelia | COL1A1 | #166200/166210/259420/166220 Osteogenesis imperfecta (AD); #114000 Caffey disease (AD) | NM_000088.3, c.4280_4283delTTGA, p.Ile1427Asnfs*98 (De novo) | Novel |
| 10 | Fetal growth restriction; cerebellar dysplasia | SBDS | #260400 Shwachman–Diamond syndrome (AR) | NM_016038.2, c.183_184delinsCT, p.Lys62* (Pat) | Reported (PMID15769891;PMID18478597;PMID15284109) |
| NM_016038.2, c.258+2T>C (Mat) | Reported (PMID15769891;PMID18478597;PMID15284109) | ||||
| 12 | Bilateral pleural effusion; hydroderma; anhydramnios | MUSK | #208150 Fetal akinesia deformation sequence 1 (AR) | NM_005592.3, c.790C>T, p.Arg264* (Mat) | Novel |
| NM_005592.3, c.1003_1006delGTTT, p.Val335Phefs*24 (Pat) | Novel | ||||
| 15 | Kidney agenesis | HNF1B | #137920 Renal cysts and diabetes syndrome (AD) | NM_000458.2, c.494G>A, p.Arg165His (De novo) | Reported (PMID24254850;PMID27838256;PMID22051731) |
| 16 | Rhabdomyomas; subependymal nodules | TSC2 | #613254 Tuberous sclerosis-2 (AD) | NM_000548.3, c.4258_4261delTCAG, p.Ser1420Glyfs*55 (De novo) | Reported (PMID26252095;PMID10533067;PMID29740858) |
| 30-1 | Rhabdomyomas; intracardiac echogenic focus | TSC2 | #613254 Tuberous sclerosis-2 (AD) | NM_000548.3, c.4762C>T, p.Gln1588* (De novo) | Reported (PMID27494029) |
| 30-2 | Rhabdomyomas; intracardiac echogenic focus | TSC2 | #613254 Tuberous sclerosis-2 (AD) | NM_000548.3, c.4762C>T, p.Gln1588* (De novo) | Reported (PMID27494029) |
| 51 # | Ventricular septal defect; choledochol cyst; ventriculomegaly; echogenic crystalline lens | GJA8 | #116200 Cataract 1, multiple types (AD) | NM_005267.4, c.593G>A, p.Arg198Gln (De novo) | Reported (PMID16604058) |
| 64 | Anhydramnios; hydroderma | RYR1 | #255320 Minicore myopathy with external ophthalmoplegia (AR) | NM_000540.2, c.6082C>T, p.Arg2028* (Pat) | Novel |
| NM_000540.2, c.165+5G>A (Mat) | Novel | ||||
| 77 | Cleft lip/palate; persistent left umbilical vein | CHD7 | #214800 CHARGE syndrome (AD) | NM_017780.3, c.2881delG, p.Glu961Serfs*16 (De novo) | Novel |
| 97 | Holoprosencephaly; agenesis of the corpus callosum | ZIC2 | #609637 Holoprosencephaly 5 (AD) | NM_007129.3, c.916G>T, p.Glu306* (De novo) | Novel |
| 104 | Fetal growth restriction | IARS1 | #617093 Growth retardation, impaired intellectual development, hypotonia, and hepatopathy (AR) | NM_013417.2 c.2420C>G, p.Pro807Arg (Mat) | Novel |
| NM_013417.2, c.2975A>G, p.Asn992Ser (Pat) | Novel | ||||
| 112 | Cleft lip/palate; small stomach; ventricular septal defect | CHD7 | #214800 CHARGE syndrome (AD) | NM_017780.3, c.7153C>T,p.Gln2385* (De novo) | Novel |
| Groups | Number of Cases | Termination of Pregnancy | Continuing Pregnancy | p Value ^ | ||
|---|---|---|---|---|---|---|
| Number of Cases | 95% CI # | Number of Cases | 95% CI # | |||
| Diagnosed | 22 (19.8%) | 21 (95.5%) | 77.2%–99.9% | 1 (4.5%) | 0.1%–22.8% | 0.00000053 |
| Undiagnosed | 89 (80.2%) | 34 (38.2%) | 28.1%–49.1% | 55 (61.8%) | 50.9%–71.9% | NA |
| Overall | 111 | 55 (49.6%) | 39.9%–59.2% | 56 (50.4%) | 40.8%–60.1% | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Yang, Z.; Sun, J.; Liu, L.; Zhou, X.; Liu, F.; Xing, Y.; Cui, S.; Xiong, S.; Liu, X.; et al. Whole Genome Sequencing in the Evaluation of Fetal Structural Anomalies: A Parallel Test with Chromosomal Microarray Plus Whole Exome Sequencing. Genes 2021, 12, 376. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030376
Zhou J, Yang Z, Sun J, Liu L, Zhou X, Liu F, Xing Y, Cui S, Xiong S, Liu X, et al. Whole Genome Sequencing in the Evaluation of Fetal Structural Anomalies: A Parallel Test with Chromosomal Microarray Plus Whole Exome Sequencing. Genes. 2021; 12(3):376. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030376
Chicago/Turabian StyleZhou, Jia, Ziying Yang, Jun Sun, Lipei Liu, Xinyao Zhou, Fengxia Liu, Ya Xing, Shuge Cui, Shiyi Xiong, Xiaoyu Liu, and et al. 2021. "Whole Genome Sequencing in the Evaluation of Fetal Structural Anomalies: A Parallel Test with Chromosomal Microarray Plus Whole Exome Sequencing" Genes 12, no. 3: 376. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030376