
Protein-Coding Genes of Helicobacter pylori Predominantly Present Purifying Selection though Many Membrane Proteins Suffer from Selection Pressure: A Proposal to Analyze Bacterial Pangenomes


Abstract
:1. Introduction
2. Materials and Methods
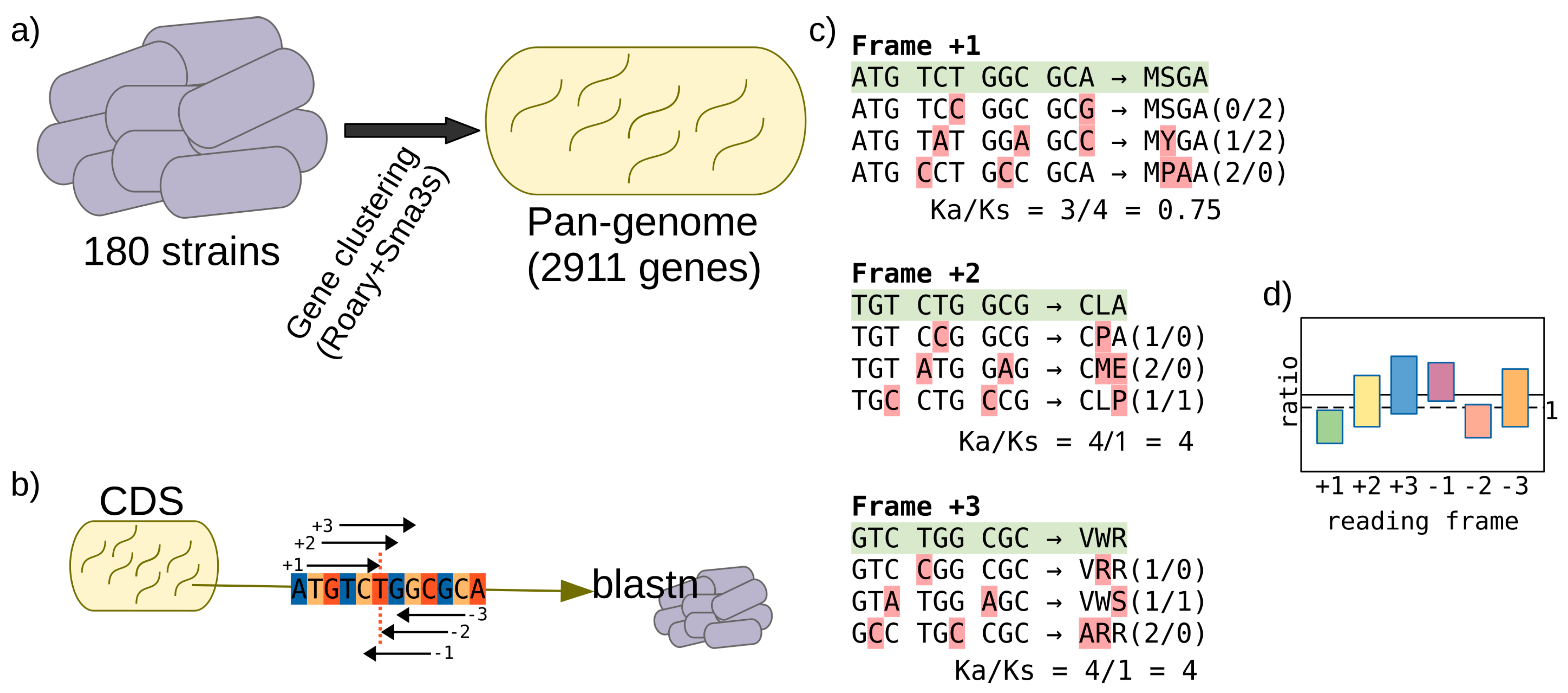
2.1. Genome Sequences, Pangenome Estimation, and Functional Annotation
2.2. Ratio Ka/Ks Calculation
2.3. Functional Enrichment
2.4. Prediction of Spurious Proteins
3. Results
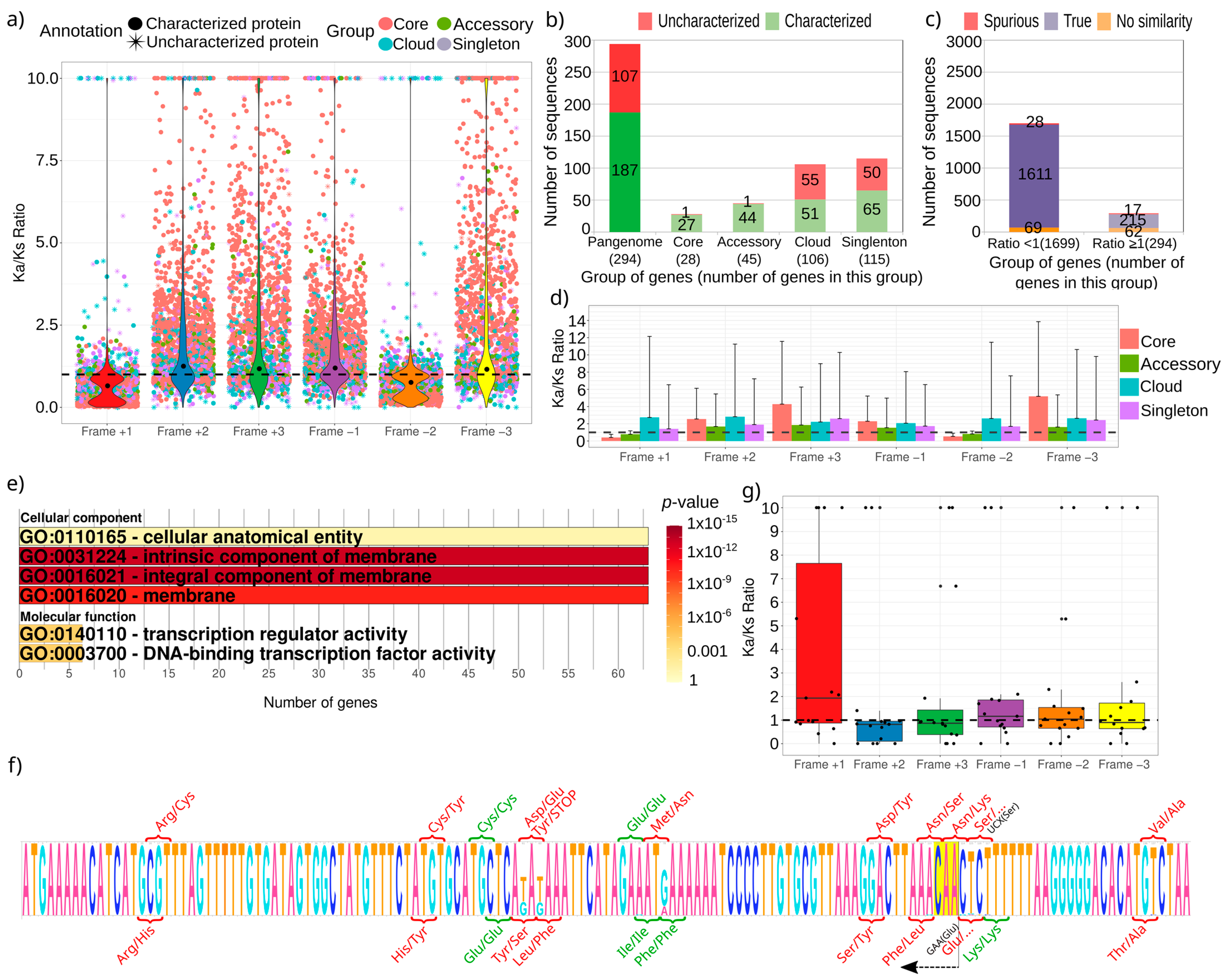
3.1. The Helicobacter Pylori Pangenome Is Twice the Number of Genes of an Independent Genome
3.2. The Ratio Ka/Ks Using the Pangenome of H. pylori Supports Purifying Selection for Most of the Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathé, C.; Sagot, M.-F.; Schiex, T.; Rouzé, P. Current Methods of Gene Prediction, Their Strengths and Weaknesses. Nucleic Acids Res. 2002, 30, 4103–4117. [Google Scholar] [CrossRef]
- Park, S.J.; Son, W.S.; Lee, B.-J. Structural Analysis of Hypothetical Proteins from Helicobacter Pylori: An Approach to Estimate Functions of Unknown or Hypothetical Proteins. Int. J. Mol. Sci. 2012, 13, 7109–7137. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.C.; Gillette, R.K.; Maglasang, M.A.; Corn, E.A.; Tai, A.K.; Lazinski, D.W.; Shanks, R.M.Q.; Kadouri, D.E.; Camilli, A. High-Throughput Analysis of Gene Function in the Bacterial Predator Bdellovibrio Bacteriovorus. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangas, E.L.; Rubio, A.; Álvarez-Marín, R.; Labrador-Herrera, G.; Pachón, J.; Pachón-Ibáñez, M.E.; Divina, F.; Pérez-Pulido, A.J. Pangenome of Acinetobacter Baumannii Uncovers Two Groups of Genomes, One of Them with Genes Involved in CRISPR/Cas Defence Systems Associated with the Absence of Plasmids and Exclusive Genes for Biofilm Formation. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.M.; Salzberg, S.L. Pan-Genomics in the Human Genome Era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Fiddes, I.T.; Diekhans, M.; Paten, B. Whole-Genome Alignment and Comparative Annotation. Annu. Rev. Anim. Biosci. 2019, 7, 41–64. [Google Scholar] [CrossRef]
- Oyanedel, D.; Labreuche, Y.; Bruto, M.; Amraoui, H.; Robino, E.; Haffner, P.; Rubio, T.; Charrière, G.M.; Le Roux, F.; Destoumieux-Garzón, D. Vibrio Splendidus O-Antigen Structure: A Trade-off between Virulence to Oysters and Resistance to Grazers. Environ. Microbiol. 2020, 22, 4264–4278. [Google Scholar] [CrossRef]
- Adrian, J.; Bonsignore, P.; Hammer, S.; Frickey, T.; Hauck, C.R. Adaptation to Host-Specific Bacterial Pathogens Drives Rapid Evolution of a Human Innate Immune Receptor. Curr. Biol. 2019, 29, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Weedall, G.D.; Polley, S.D.; Conway, D.J. Gene-Specific Signatures of Elevated Non-Synonymous Substitution Rates Correlate Poorly across the Plasmodium Genus. PLoS ONE 2008, 3, e2281. [Google Scholar] [CrossRef]
- Guéguen, L.; Duret, L. Unbiased Estimate of Synonymous and Nonsynonymous Substitution Rates with Nonstationary Base Composition. Mol. Biol. Evol. 2018, 35, 734–742. [Google Scholar] [CrossRef] [Green Version]
- Jordan, I.K.; Rogozin, I.B.; Wolf, Y.I.; Koonin, E.V. Microevolutionary Genomics of Bacteria. Theor. Popul. Biol. 2002, 61, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Algood, H.M.S.; Cover, T.L. Helicobacter Pylori Persistence: An Overview of Interactions between H. Pylori and Host Immune Defenses. Clin. Microbiol. Rev. 2006, 19, 597–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, R.A.; Eccersley, A.J.; Hardie, J.M. Epidemiology of Helicobacter Pylori: Acquisition, Transmission, Population Prevalence and Disease-to-Infection Ratio. Br. Med. Bull. 1998, 54, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressmann, H.; Linz, B.; Ghai, R.; Pleissner, K.-P.; Schlapbach, R.; Yamaoka, Y.; Kraft, C.; Suerbaum, S.; Meyer, T.F.; Achtman, M. Gain and Loss of Multiple Genes during the Evolution of Helicobacter Pylori. PLoS Genet. 2005, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- Oleastro, M.; Ménard, A. The Role of Helicobacter Pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology 2013, 2, 1110–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alm, R.A.; Bina, J.; Andrews, B.M.; Doig, P.; Hancock, R.E.; Trust, T.J. Comparative Genomics of Helicobacter Pylori: Analysis of the Outer Membrane Protein Families. Infect. Immun. 2000, 68, 4155–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmiela, M.; Walczak, N.; Rudnicka, K. Helicobacter Pylori Outer Membrane Vesicles Involvement in the Infection Development and Helicobacter Pylori-Related Diseases. J. Biomed. Sci. 2018, 25, 78. [Google Scholar] [CrossRef] [PubMed]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.-E.; Arnqvist, A.; Vikström, E. Helicobacter Pylori Vesicles Carrying CagA Localize in the Vicinity of Cell-Cell Contacts and Induce Histone H1 Binding to ATP in Epithelial Cells. FEMS Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Casimiro-Soriguer, C.S.; Muñoz-Mérida, A.; Pérez-Pulido, A.J. Sma3s: A Universal Tool for Easy Functional Annotation of Proteomes and Transcriptomes. Proteomics 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved Scoring of Functional Groups from Gene Expression Data by Decorrelating GO Graph Structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Höps, W.; Jeffryes, M.; Bateman, A. Gene Unprediction with Spurio: A Tool to Identify Spurious Protein Sequences. F1000Res 2018, 7, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, O.; Salama, N.R. The Helicobacter Pylori HpyAXII Restriction-Modification System Limits Exogenous DNA Uptake by Targeting GTAC Sites but Shows Asymmetric Conservation of the DNA Methyltransferase and Restriction Endonuclease Components. Nucleic Acids Res. 2008, 36, 6893–6906. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Ishiguro, K.; Watanabe, O.; Miyake, N.; Kato, T.; Hibi, S.; Mimura, S.; Nakamura, M.; Miyahara, R.; Ohmiya, N.; et al. Restriction-Modification Systems May Be Associated with Helicobacter Pylori Virulence. J. Gastroenterol Hepatol. 2010, 25 Suppl. 1, S95–S98. [Google Scholar] [CrossRef]
- Vale, F.F.; Mégraud, F.; Vítor, J.M. Geographic Distribution of Methyltransferases of Helicobacter Pylori: Evidence of Human Host Population Isolation and Migration. BMC Microbiol. 2009, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangpanwimon, K.; Sottisuporn, J.; Mittraparp-Arthorn, P.; Ueaphatthanaphanich, W.; Rattanasupar, A.; Pourcel, C.; Vuddhakul, V. CRISPR-like Sequences in Helicobacter Pylori and Application in Genotyping. Gut Pathog. 2017, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- García-Zea, J.A.; de la Herrán, R.; Robles Rodríguez, F.; Navajas-Pérez, R.; Ruiz Rejón, C. Detection and Variability Analyses of CRISPR-like Loci in the H. Pylori Genome. PeerJ 2019, 7. [Google Scholar] [CrossRef]
- Tunca, S.; Barreiro, C.; Coque, J.-J.R.; Martín, J.F. Two Overlapping Antiparallel Genes Encoding the Iron Regulator DmdR1 and the Adm Proteins Control Siderophore [Correction of Sedephore] and Antibiotic Biosynthesis in Streptomyces Coelicolor A3(2). FEBS J. 2009, 276, 4814–4827. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Bechtel, N.; Witting, M.A.; Simon, S.; Schmitt-Kopplin, P.; Keim, D.; Scherer, S.; Neuhaus, K. Phenotype of HtgA (MbiA), a Recently Evolved Orphan Gene of Escherichia Coli and Shigella, Completely Overlapping in Antisense to YaaW. FEMS Microbiol. Lett. 2014, 350, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Mir, K.; Schober, S. Selection Pressure in Alternative Reading Frames. PLoS ONE 2014, 9, e108768. [Google Scholar] [CrossRef]
- Aguileta, G.; Refrégier, G.; Yockteng, R.; Fournier, E.; Giraud, T. Rapidly Evolving Genes in Pathogens: Methods for Detecting Positive Selection and Examples among Fungi, Bacteria, Viruses and Protists. Infect. Genet. Evol. 2009, 9, 656–670. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Weissman, S.J.; Minin, V.N.; Russo, T.A.; Dykhuizen, D.E.; Sokurenko, E.V. High Frequency of Hotspot Mutations in Core Genes of Escherichia Coli Due to Short-Term Positive Selection. Proc. Natl. Acad. Sci. USA 2009, 106, 12412–12417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Chi, P.B.; Minin, V.N.; Berg, D.E.; Sokurenko, E.V. Recombination-Independent Rapid Convergent Evolution of the Gastric Pathogen Helicobacter Pylori. BMC Genom. 2018, 19, 835. [Google Scholar] [CrossRef]
- Kim, J.S.; Chang, J.H.; Seo, W.Y.; Yu, G.J.; Chung, S.I.; Yum, J.S. Cloning and Characterization of a 22 KDa Outer-Membrane Protein (Omp22) from Helicobacter Pylori. Mol. Cells 2000, 10, 633–641. [Google Scholar] [CrossRef]
- López-Vidal, Y.; Ponce-de-León, S.; Castillo-Rojas, G.; Barreto-Zúñiga, R.; Torre-Delgadillo, A. High Diversity of VacA and CagA Helicobacter Pylori Genotypes in Patients with and without Gastric Cancer. PLoS ONE 2008, 3, e3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Yamakawa, A.; Okuda, T.; Ohtani, M.; Suto, H.; Ito, Y.; Yamazaki, Y.; Keida, Y.; Higashi, H.; Hatakeyama, M.; et al. Distinct Diversity of VacA, CagA, and CagE Genes of Helicobacter Pylori Associated with Peptic Ulcer in Japan. J. Clin. Microbiol. 2005, 43, 3906–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene | Group | Ratio(+1) | Ratio(-2) | Gene | Group | Ratio(+1) | Ratio(-2) | Gene | Group | Ratio(+1) | Ratio(-2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| alpB | Core | - | - | hopH | Core | 0.905 | HPAKL86_04155 | Singleton | - | - | |
| babB | Singleton | - | - | hopI | Core | 0.872 | 0.939 | HPAKL86_05700 | Cloud | 0.275 | 0.326 |
| babC | Singleton | - | - | hopK | Core | 0.903 | 1.272 | HPY1089_07465 | Accessory | 0.876 | 0.870 |
| bamD | Core | 0.698 | 0.857 | hopL | Core | 0.311 | 0.893 | jhp_0663 | Core | 0.877 | 1.065 |
| fecA | Core | - | - | hopM | Singleton | 1.001 | - | jhp_1360 | Core | 0.960 | |
| fecA2 | Accessory | 1.139 | hopP | Accessory | - | - | omp13 | Singleton | 0.125 | 0.243 | |
| frpB | Core | 0.732 | 0.907 | hopZ | Accessory | - | - | omp17 | Singleton | 0.042 | 0.219 |
| hefD | Core | 0.114 | 0.240 | horA | Accessory | - | 1.746 | omp21 | Singleton | 0.255 | 0.389 |
| hefG | Core | 0.228 | 0.334 | horB | Core | 0.039 | 0.126 | omp22 | Singleton | 1.225 | - |
| hofB | Core | 1.253 | 1.742 | horC | Core | 0.124 | 0.244 | omp24 | Singleton | 0.870 | 0.967 |
| hofC | Core | 0.115 | 0.233 | horE | Core | 0.041 | 0.218 | omp25 | Singleton | 0.922 | 0.911 |
| hofE | Core | 0.370 | 0.536 | horF | Core | 0.102 | 0.182 | omp26 | Singleton | 0.186 | 0.370 |
| hofF | Core | 0.080 | 0.205 | horG | Core | 0.238 | 0.360 | omp33 | Singleton | 0.164 | 0.302 |
| hofG | Core | 0.753 | 0.889 | horH | Core | 0.245 | 0.288 | omp4 | Singleton | 0.316 | 0.367 |
| homC | Core | - | - | horI | Core | 0.210 | 0.487 | omp5 | Singleton | - | - |
| homD | Core | 1.010 | 0.830 | horJ | Core | 0.125 | 0.216 | ompP1 | Core | 0.799 | 0.989 |
| hopA | Core | 0.907 | 0.716 | horL | Core | - | 1.066 | sabA | Singleton | - | - |
| hopD | Core | 0.661 | HP_1426 | Accessory | 0.928 | - | sabB | Singleton | 0.346 | 0.537 | |
| hopE | Core | 0.088 | 0.132 | hp908_0703 | Core | 0.050 | 0.195 | unknown40 | Accessory | 1.029 | 1.009 |
| hopF | Core | 0.270 | 0.352 | hp908_1474 | Core | 0.169 | 0.273 | vlpC | Accessory | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio, A.; Pérez-Pulido, A.J. Protein-Coding Genes of Helicobacter pylori Predominantly Present Purifying Selection though Many Membrane Proteins Suffer from Selection Pressure: A Proposal to Analyze Bacterial Pangenomes. Genes 2021, 12, 377. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030377
Rubio A, Pérez-Pulido AJ. Protein-Coding Genes of Helicobacter pylori Predominantly Present Purifying Selection though Many Membrane Proteins Suffer from Selection Pressure: A Proposal to Analyze Bacterial Pangenomes. Genes. 2021; 12(3):377. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030377
Chicago/Turabian StyleRubio, Alejandro, and Antonio J. Pérez-Pulido. 2021. "Protein-Coding Genes of Helicobacter pylori Predominantly Present Purifying Selection though Many Membrane Proteins Suffer from Selection Pressure: A Proposal to Analyze Bacterial Pangenomes" Genes 12, no. 3: 377. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030377