
Shared Ancestry and Signatures of Recent Selection in Gotland Sheep

 , ,
, , 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Data Quality Control
2.2. Population Genetic Structure Analysis
2.2.1. Principal Component (PC) Analysis
2.2.2. Individual Ancestry Coefficients
2.3. Identifying Genomic Regions Showing Recent Selection Signatures
2.3.1. Bayesian Population Differentiation (FST)
2.3.2. Integrated Haplotype Homozygosity Score (iHS)
2.3.3. Identification of Genes Located in the Regions Showing Selection Signature
3. Results and Discussion
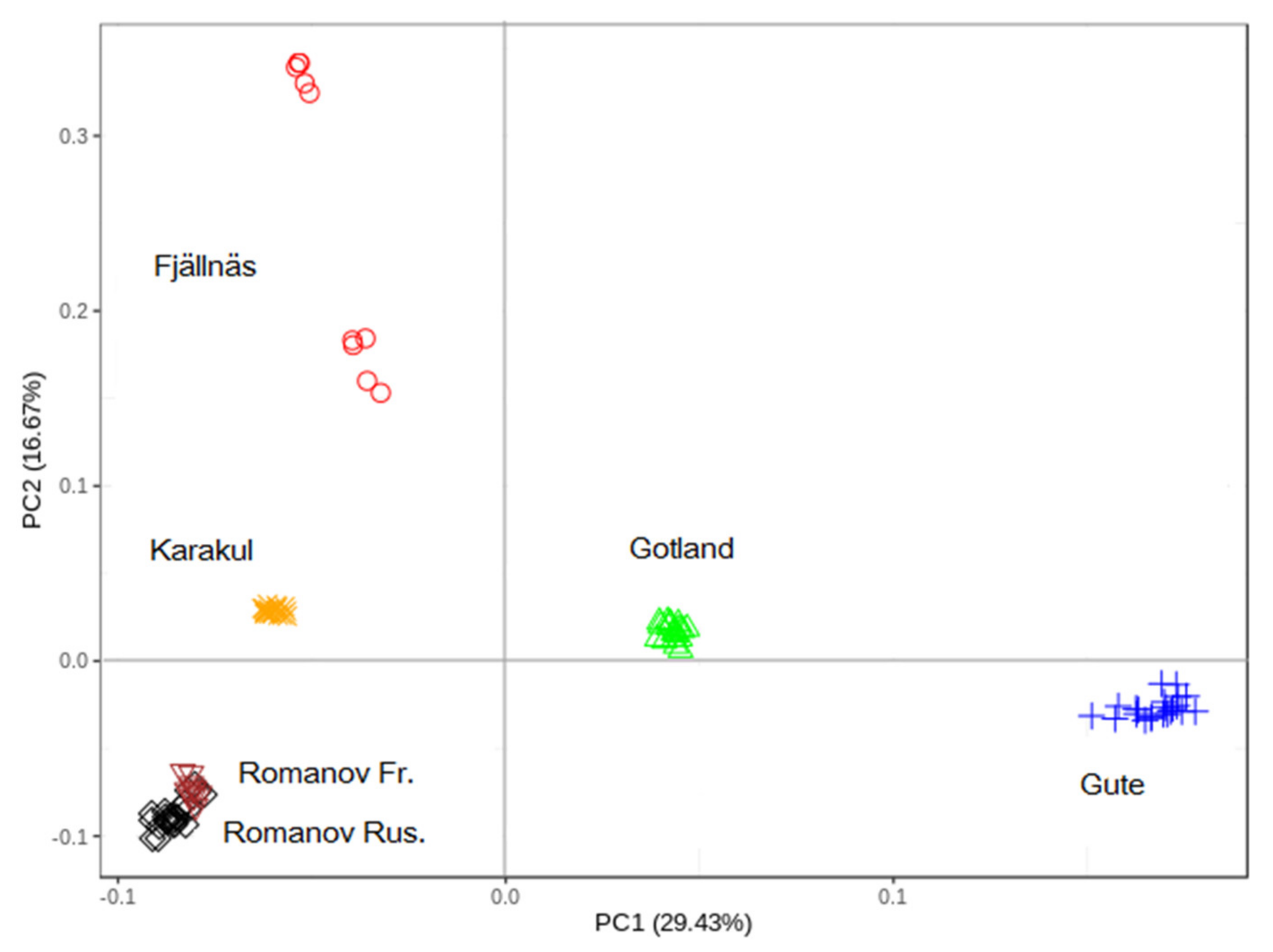
3.1. Principal Component (PC) Analysis
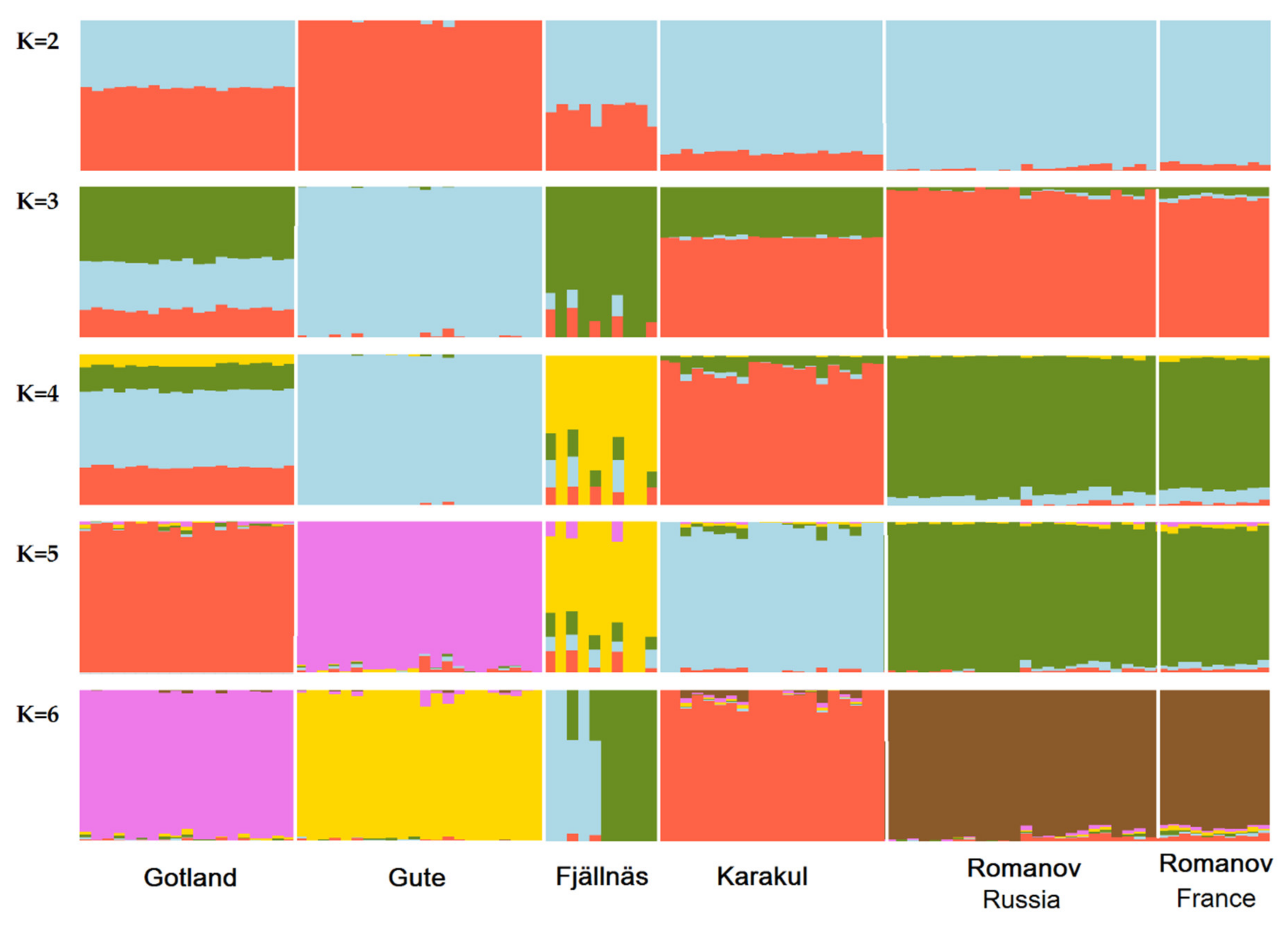
3.2. Population Admixture
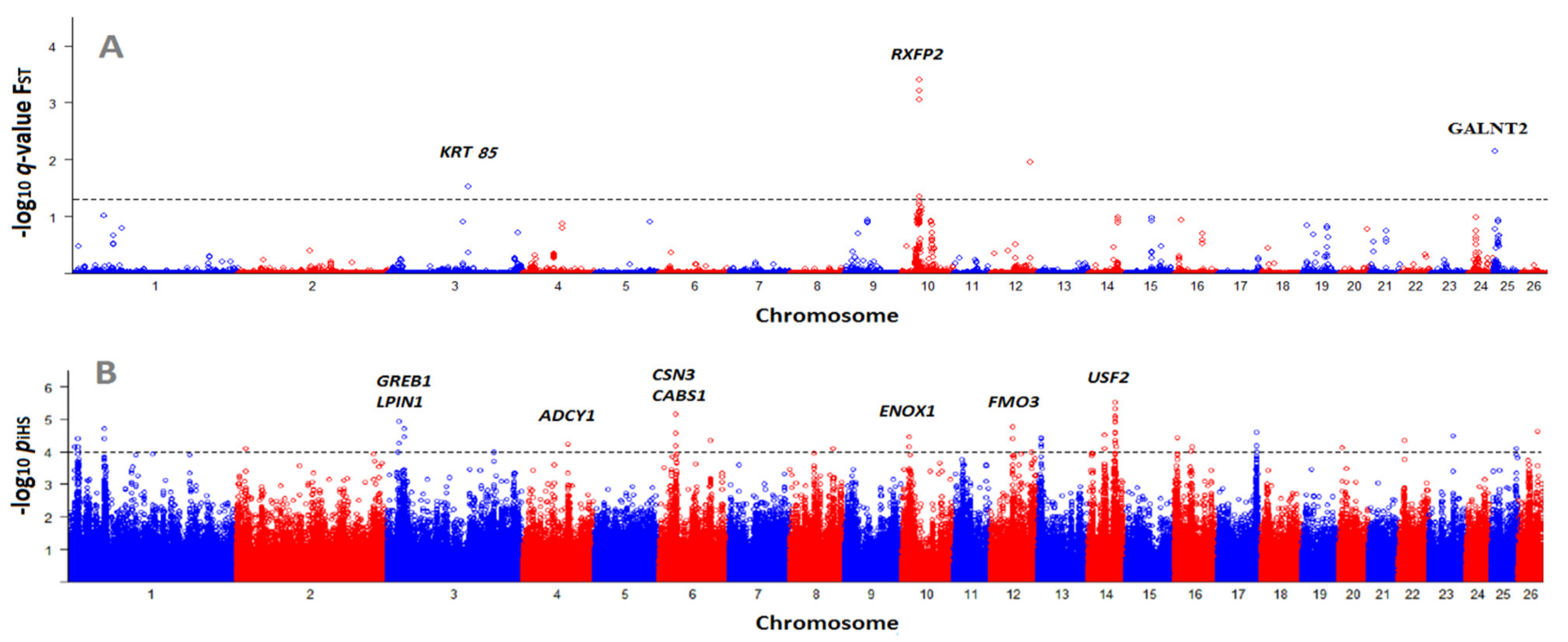
3.3. Identification of Selection Signatures with BayeScan and iHS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rochus, C.M.; Jonas, E.; Johansson, A.M. Population structure of five native sheep breeds of Sweden estimated with high density SNP genotypes. BMC Genet. 2020, 21, 27. [Google Scholar] [CrossRef]
- Näsholm, A. Genetic study on pelt quality traits in the Gotland sheep breed. Acta Agric. Scand. Sect. A Anim. Sci. 2005, 55, 57–65. [Google Scholar] [CrossRef]
- Van Genderen, C.; Okamura, R.M.; Farinas, I.; Quo, R.G.; Parslow, T.G.; Bruhn, L.; Grosschedl, R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994, 8, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- Butler, L.; D’Orazio, R.; Ahlén, K. Review of the Gotland pelt sheep industry. Small Rumin. Res. 1993, 10, 33–44. [Google Scholar] [CrossRef]
- Dýrmundsson, Ó.R.; Niżnikowski, R. North European short-tailed breeds of sheep: A review. Animal 2010, 4, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Safdarian, M.; Kafi, M.; Hashemi, M. Reproductive Performance of Karakul Ewes Following Different Oestrous Synchronisation Treatments Outside the Natural Breeding Season. S. Afr. J. Anim. Sci. 2006, 36, 229–234. [Google Scholar]
- Rochus, C.M.; Johansson, A.M. Estimation of genetic diversity in Gute sheep: Pedigree and microsatellite analyses of an ancient Swedish breed. Hereditas 2017, 154, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoreishifar, S.M.; Eriksson, S.; Johansson, A.M.; Khansefid, M.; Moghaddaszadeh-Ahrabi, S.; Parna, N.; Davoudi, P.; Javanmard, A. Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds. Genet. Sel. Evol. 2020, 52, 52. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, J.J.D.S.; Paiva, S.R.; McManus, C.M.; Caetano, A.R.; Kijas, J.W.; Facó, O.; Azevedo, H.C.; De Araujo, A.M.; De Souza, C.J.H.; Yamagishi, M.E.B.; et al. Genome-wide search for signatures of selection in three major Brazilian locally adapted sheep breeds. Livest. Sci. 2017, 197, 36–45. [Google Scholar] [CrossRef]
- Kijas, J.W. Haplotype-based analysis of selective sweeps in sheep. Genome 2014, 57, 433–437. [Google Scholar] [CrossRef]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 2019, 20, 294. [Google Scholar] [CrossRef] [PubMed]
- Rochus, C.M.; Tortereau, F.; Plisson-Petit, F.; Restoux, G.; Moreno-Romieux, C.; Tosser-Klopp, G.; Servin, B. Revealing the selection history of adaptive loci using genome-wide scans for selection: An example from domestic sheep. BMC Genom. 2018, 19, 71. [Google Scholar] [CrossRef] [Green Version]
- Signer-Hasler, H.; Burren, A.; Ammann, P.; Drögemüller, C.; Flury, C. Runs of Homozygosity and Signatures of Selection: A Comparison among Eight Local Swiss Sheep Breeds. Anim. Genet. 2019, 50, 512–525. [Google Scholar] [CrossRef]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [Green Version]
- Qanbari, S.; Pimentel, E.C.G.; Tetens, J.; Thaller, G.; Lichtner, P.; Sharifi, A.R.; Simianer, H. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim. Genet. 2010, 41, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishifar, S.M.; Moradi-Shahrbabak, H.; Fallahi, M.H.; Sarghale, A.J.; Moradi-Shahrbabak, M.; Abdollahi-Arpanahi, R.; Khansefid, M. Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo, Bubalus bubalis. BMC Genet. 2020, 21, 16. [Google Scholar] [CrossRef] [Green Version]
- Salek Ardestani, S.; Aminafshar, M.; Maryam, M.B.Z.B.; Banabazi, M.H.; Sargolzaei, M.; Miar, Y. Signatures of selection analysis using whole-genome sequence data reveals novel candidate genes for pony and light horse types. Genome 2020, 63, 387–396. [Google Scholar] [CrossRef]
- Bertolini, F.; The ADAPTMAP Consortium; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhière, I.; Crisà, A.; Catillo, G.; et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Sel. Evol. 2018, 50, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Edea, Z.; Kim, K.-S. A whole genomic scan to detect selection signatures between Berkshire and Korean native pig breeds. J. Anim. Sci. Technol. 2014, 56, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salek Ardestani, S.; Aminafshar, M.; Maryam, M.B.Z.B.; Banabazi, M.H.; Sargolzaei, M.; Miar, Y. Whole-Genome Signatures of Selection in Sport Horses Revealed Selection Footprints Related to Musculoskeletal System Development Processes. Animals 2019, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.Y.; Toghiani, S.; Ling, A.; Aggrey, S.E.; Rekaya, R. High Density Marker Panels, SNPs Prioritizing and Accuracy of Genomic Selection. BMC Genet. 2018, 19, 4. [Google Scholar]
- Chang, L.-Y.; Toghiani, S.; Hay, E.H.; Aggrey, S.E.; Rekaya, R. A Weighted Genomic Relationship Matrix Based on Fixation Index (FST) Prioritized SNPs for Genomic Selection. Genes 2019, 10, 922. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 11 March 2021).
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Frichot, E.; Mathieu, F.; Trouillon, T.; Bouchard, G.; François, O. Fast and Efficient Estimation of Individual Ancestry Coefficients. Genetics 2014, 196, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O.E. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaneau, O.; Zagury, J.F.; Marchini, J. Improved Whole-Chromosome Phasing for Disease and Population Genetic Studies. Nat. Methods 2013, 10, 5. [Google Scholar] [CrossRef]
- Petit, M.; Astruc, J.-M.; Sarry, J.; Drouilhet, L.; Fabre, S.; Moreno, C.R.; Servin, B. Variation in Recombination Rate and Its Genetic Determinism in Sheep Populations. Genetics 2017, 207, 767–784. [Google Scholar] [CrossRef] [Green Version]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasprzyk, A. BioMart: Driving a paradigm change in biological data management. Database 2011, 2011, bar049. [Google Scholar] [CrossRef] [PubMed]
- Ekman, S. Das Gotlandshaf (the Gotland Sheep). Zeitschrift Säugetierkunde 1964, 23, 123–145. [Google Scholar]
- Pan, Z.; Li, S.; Liu, Q.; Wang, Z.; Zhou, Z.; Di, R.; Miao, B.; Hu, W.; Wang, X.; Hu, X.; et al. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 2018, 7, 19. [Google Scholar] [CrossRef]
- Haehling, M.B.; Cruvinel, G.G.; Toscano, J.H.; Giraldelo, L.A.; Santos, I.B.; Esteves, S.N.; Benavides, M.V.; Júnior, W.B.; Niciura, S.C.; Chagas, A.C.S. Four single nucleotide polymorphisms (SNPs) are associated with resistance and resilience to Haemonchus contortus in Brazilian Morada Nova sheep. Veter Parasitol. 2020, 279, 109053. [Google Scholar] [CrossRef]
- Guo, T.; Yin, R.-X.; Huang, F.; Yao, L.-M.; Lin, W.-X.; Pan, S.-L. Association between the DOCK7, PCSK9 and GALNT2 Gene Polymorphisms and Serum Lipid levels. Sci. Rep. 2016, 6, 19079. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, L.; Gao, H.; Wu, Y.; Gao, X.; Zhang, L.; Zhu, B.; Song, Y.; Bao, J.; Li, J.; et al. Detection of candidate genes for growth and carcass traits using genome-wide association strategy in Chinese Simmental beef cattle. Anim. Prod. Sci. 2018, 58, 224. [Google Scholar] [CrossRef]
- Yu, Z.; Gordon, S.W.; Nixon, A.J.; Bawden, C.S.; Rogers, M.A.; Wildermoth, J.E.; Maqbool, N.J.; Pearson, A.J. Expression patterns of keratin intermediate filament and keratin associated protein genes in wool follicles. Differentiation 2009, 77, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Zhou, H.; Forrest, R.H.; Gong, H.; Hodge, S.; Hickford, J.G. Polymorphism of KRT83 and its association with selected wool traits in Merino-cross lambs. Small Rumin. Res. 2017, 155, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Liu, G.; Liu, Y.; Xu, Q.; Zhang, M.; Fang, M. Transcriptome Profile at Different Physiological Stages Reveals Potential Mode for Curly Fleece in Chinese Tan Sheep. PLoS ONE 2013, 8, e71763. [Google Scholar] [CrossRef] [Green Version]
- Moradian, H.; Koshkoiyeh, A.E.; Mohammadabadi, M.; Fozi, M.A. Whole genome detection of recent selection signatures in Sarabi cattle: A unique Iranian taurine breed. Genes Genom. 2020, 42, 203–215. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Q.; Di, R.; Hu, W.; Wang, X.; He, X.; Ma, L.; Chu, M. Single nucleotide polymorphisms in BMP2 and BMP7 and the association with litter size in Small Tail Han sheep. Anim. Reprod. Sci. 2019, 204, 183–192. [Google Scholar] [CrossRef]
- Lai, F.-N.; Zhai, H.-L.; Cheng, M.; Ma, J.-Y.; Cheng, S.-F.; Ge, W.; Zhang, G.-L.; Wang, J.-J.; Zhang, R.-Q.; Wang, X.; et al. Whole-genome scanning for the litter size trait associated genes and SNPs under selection in dairy goat (Capra hircus). Sci. Rep. 2016, 6, 38096. [Google Scholar] [CrossRef]
- Tang, J.; Hu, W.; Chen, S.; Di, R.; Liu, Q.; Wang, X.; He, X.; Gan, S.; Zhang, X.; Zhang, J.; et al. The genetic mechanism of high prolificacy in small tail han sheep by comparative proteomics of ovaries in the follicular and luteal stages. J. Proteom. 2019, 204, 103394. [Google Scholar] [CrossRef] [PubMed]
- Kongsuwan, K.; Knox, M.; Allingham, P.; Pearson, R.; Dalrymple, B. The effect of combination treatment with trenbolone acetate and estradiol-17β on skeletal muscle expression and plasma concentrations of oxytocin in sheep. Domest. Anim. Endocrinol. 2012, 43, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Klopp, C.; Tosser-Klopp, G.; Arranz, J.J. Variant discovery in the sheep milk transcriptome using RNA sequencing. BMC Genom. 2017, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, H.; Gong, H.; He, J.; Luo, Y.; Hickford, J.G.H.; Hu, J.; Wang, J.; Liu, X.; Li, S. Variation in the Lipin 1 Gene Is Associated with Birth Weight and Selected Carcass Traits in New Zealand Romney Sheep. Animals 2020, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, H.; Hickford, J.G.H.; Li, S.; Wang, J.; Liu, X.; Hu, J.; Luo, Y. Variation in the yak lipin-1 gene and its association with milk traits. J. Dairy Res. 2020, 87, 166–169. [Google Scholar] [CrossRef]
- Yudin, N.; Larkin, D.M. Shared Signatures of Selection Related to Adaptation and Acclimation in Local Cattle and Sheep Breeds from Russia. Russ. J. Genet. 2019, 55, 1008–1014. [Google Scholar] [CrossRef]
- Chen, H.; Lan, X.Y.; Lei, C.Z.; Pan, C.Y.; Zhang, R.F.; Zhang, Y.D.; Li, R.B. Association of Csn3 and Csn1s2 Genes with Litter Size in Chinese Xinong Saanen Dairy Goat. In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Minas Gerais, Brazil, 13–18 August 2006. [Google Scholar]
- Wang, K.; Yan, H.; Xu, H.; Yang, Q.; Zhang, S.; Pan, C.; Chen, H.; Zhu, H.; Liu, J.; Qu, L.; et al. A novel indel within goat casein alpha S1 gene is significantly associated with litter size. Gene 2018, 671, 161–169. [Google Scholar] [CrossRef]
- Lan, X.Y.; Chen, H.; Pan, C.Y.; Li, R.B.; Li, X.C.; Fang, X.T. Relationship between Polymorphisms of Csn3, Csn1s2 and Β-Lg Genes and Litter Sizes of Xinong Saanen Dairy Goat. Sci. Agric. Sin. 2005, 11, 2333–2338. [Google Scholar]
- Sell-Kubiak, E.; Duijvesteijn, N.; Lopes, M.S.; Janss, L.L.G.; Knol, E.F.; Bijma, P.; Mulder, H.A. Genome-wide association study reveals novel loci for litter size and its variability in a Large White pig population. BMC Genom. 2015, 16, 1049. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, G.; Xu, X.; Geng, R.; Zhou, J.; Yang, Y.; Yang, Z.; Chen, Y. Transcriptome profile analysis of adipose tissues from fat and short-tailed sheep. Gene 2014, 549, 252–257. [Google Scholar] [CrossRef]
- Bai, M.; Sun, L.; Zhao, J.; Xiang, L.; Cheng, X.; Li, J.; Jia, C.; Jiang, H. Histological analysis and identification of spermatogenesis-related genes in 2-, 6-, and 12-month-old sheep testes. Naturwissenschaften 2017, 104, 84. [Google Scholar] [CrossRef]
- Grochowska, E.; Borys, B.; Grześkowiak, E.; Mroczkowski, S. Effect of the calpain small subunit 1 gene (CAPNS1) polymorphism on meat quality traits in sheep. Small Rumin. Res. 2017, 150, 15–21. [Google Scholar] [CrossRef]
- Machado, A.L.; Meira, A.N.; Muniz, E.N.; Azevedo, H.C.; Coutinho, L.L.; Mourão, G.B.; Pedrosa, V.B.; Pinto, L.F.B. Single Loci and Haplotypes in CAPN1 and CAST Genes are Associated with Growth, Biometrics, and in Vivo Carcass Traits in Santa Inês Sheep. Ann. Anim. Sci. 2020, 20, 465–483. [Google Scholar] [CrossRef]
- Feng, X.; Li, F.; Wang, F.; Zhang, G.; Pang, J.; Ren, C.; Zhang, T.; Yang, H.; Wang, Z.; Zhang, Y. Genome-wide differential expression profiling of mRNAs and lncRNAs associated with prolificacy in Hu sheep. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Rovadoscki, G.A.; Pertile, S.F.N.; Alvarenga, A.B.; Cesar, A.S.M.; Pértille, F.; Petrini, J.; Franzo, V.; Soares, W.V.B.; Morota, G.; Spangler, M.L.; et al. Estimates of genomic heritability and genome-wide association study for fatty acids profile in Santa Inês sheep. BMC Genom. 2018, 19, 375. [Google Scholar] [CrossRef]
- Moioli, B.; Steri, R.; Marchitelli, C.; Catillo, G.; Buttazzoni, L. Genetic parameters and genome-wide associations of twinning rate in a local breed, the Maremmana cattle. Anim. 2017, 11, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J.; Davis, P. Mitochondrial Signaling and Fertilization. Mol. Hum. Reprod. 2007, 13, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Catalano, R.; Critchley, H.; Heikinheimo, O.; Baird, D.; Hapangama, D.; Sherwin, J.; Charnock-Jones, D.; Smith, S.; Sharkey, A. Mifepristone induced progesterone withdrawal reveals novel regulatory pathways in human endometrium. Mol. Hum. Reprod. 2007, 13, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of Demographic History and Neutral Parameterization on the Performance of Fst Outlier Tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Breed | Sample | Characteristic | Cite |
|---|---|---|---|
| Gute | Sweden (n = 22) | Primitive breed native to the island of Gotland (Sweden), the horned phenotype in both rams and ewes, bred for conservation purposes, short tail | [1] |
| Gotland | Sweden (n = 19) | Native to the island of Gotland (Sweden), split from the Gute sheep breed, probably crossed with other breeds, including Karakul and Romanov, polled phenotype, bred for commercial purposes (e.g., pelt quality), the fleece is curly | [1] |
| Fjällnäs | Sweden (n = 10) | Accepted officially as a breed in 2011, from northern Sweden, very small effective population size, has shared ancestry with Gotland breed | [1] |
| Karakul | Russia (n = 20) | Horned and polled phenotypes, fat tail, breed raised for fleece and meat | [13] |
| Romanov | Russia (n = 24) France (n = 10) | Native breed from Russia, known for its high prolificacy, lambing of litters, early sexually maturing age, year-round breeding | [13,14] |
| SNP Position | q-Value | Gene | Gene Position | Trait | Reference |
|---|---|---|---|---|---|
| 10:29,455,959 | 0.0008 | RXFP2 | within | Horned/polled phenotypes | [36] |
| 12:63,873,649 | 0.0100 | EDEM3 | within | Gastrointestinal nematodes | [37] |
| 25:1,759,489 | 0.0100 | GALNT2 | 96.7 Kb up | Growth and carcass traits | [38,39] |
| 3:133,648,712 | 0.0300 | KRT85 | - | Wool quality and quantity | [40] |
| SNP Position | piHS1 | Gene Name | Gene Position 2 | Trait | Reference |
|---|---|---|---|---|---|
| 14:45,845,449 | 4.95 | COX7A1 | 82.8 Kb up | Prolificacy | [46] |
| 3:20,370,238 | 4.28 | GREB1 | 6.4 Kb up | Muscle growth | [47] |
| 3:20,576,558 | 4.94 | LPIN1 | Within | Milk fat, growth, carcass | [48,49,50] |
| 3:28,665,400 | 4.72 | APOB | 6.4 Kb up | Cold acclimation | [51] |
| 4:76,392,859 | 4.24 | ADCY1 | 61.4 Kb down | Fecundity | [45] |
| 6:85,405,971 | 4.34 | CSN3 | 89.1 Kb down | Litter size | [52,53,54] |
| 10:14,096,224 | 4.47 | ENOX1 | Within | Litter size | [55] |
| 12:36,960,517 | 4.41 | FMO3 | 132.2 Kb down | Fat deposition | [56] |
| 12:37,377,415 | 4.77 | MYOC | 112.1 Kb down | Spermatogenesis | [57] |
| 14:45,201,813 | 4.61 | USF2 | 107.9 Kb down | Litter size | [44] |
| 14:45,845,449 | 4.95 | CAPNS1 | 75.8 Kb up | Growth, meat quality | [58,59] |
| 14:45,538,588 | 4.27 | TYROBP | 175 Kb up | Prolificacy | [60] |
| 16:29,966,767 | 4.15 | MRPS30 | 75.5 Kb up | Fatty acid profile | [61] |
| 23:40,285,834 | 4.50 | ARHGAP28 | 13.6 Kb down | Twinning rate | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoreishifar, S.M.; Rochus, C.M.; Moghaddaszadeh-Ahrabi, S.; Davoudi, P.; Salek Ardestani, S.; Zinovieva, N.A.; Deniskova, T.E.; Johansson, A.M. Shared Ancestry and Signatures of Recent Selection in Gotland Sheep. Genes 2021, 12, 433. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030433
Ghoreishifar SM, Rochus CM, Moghaddaszadeh-Ahrabi S, Davoudi P, Salek Ardestani S, Zinovieva NA, Deniskova TE, Johansson AM. Shared Ancestry and Signatures of Recent Selection in Gotland Sheep. Genes. 2021; 12(3):433. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030433
Chicago/Turabian StyleGhoreishifar, Seyed Mohammad, Christina Marie Rochus, Sima Moghaddaszadeh-Ahrabi, Pourya Davoudi, Siavash Salek Ardestani, Natalia A. Zinovieva, Tatiana E. Deniskova, and Anna M. Johansson. 2021. "Shared Ancestry and Signatures of Recent Selection in Gotland Sheep" Genes 12, no. 3: 433. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030433