
DNA Methylation at ATP11A cg11702988 Is a Biomarker of Lung Disease Severity in Cystic Fibrosis: A Longitudinal Study

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cohorts and Samples Collection
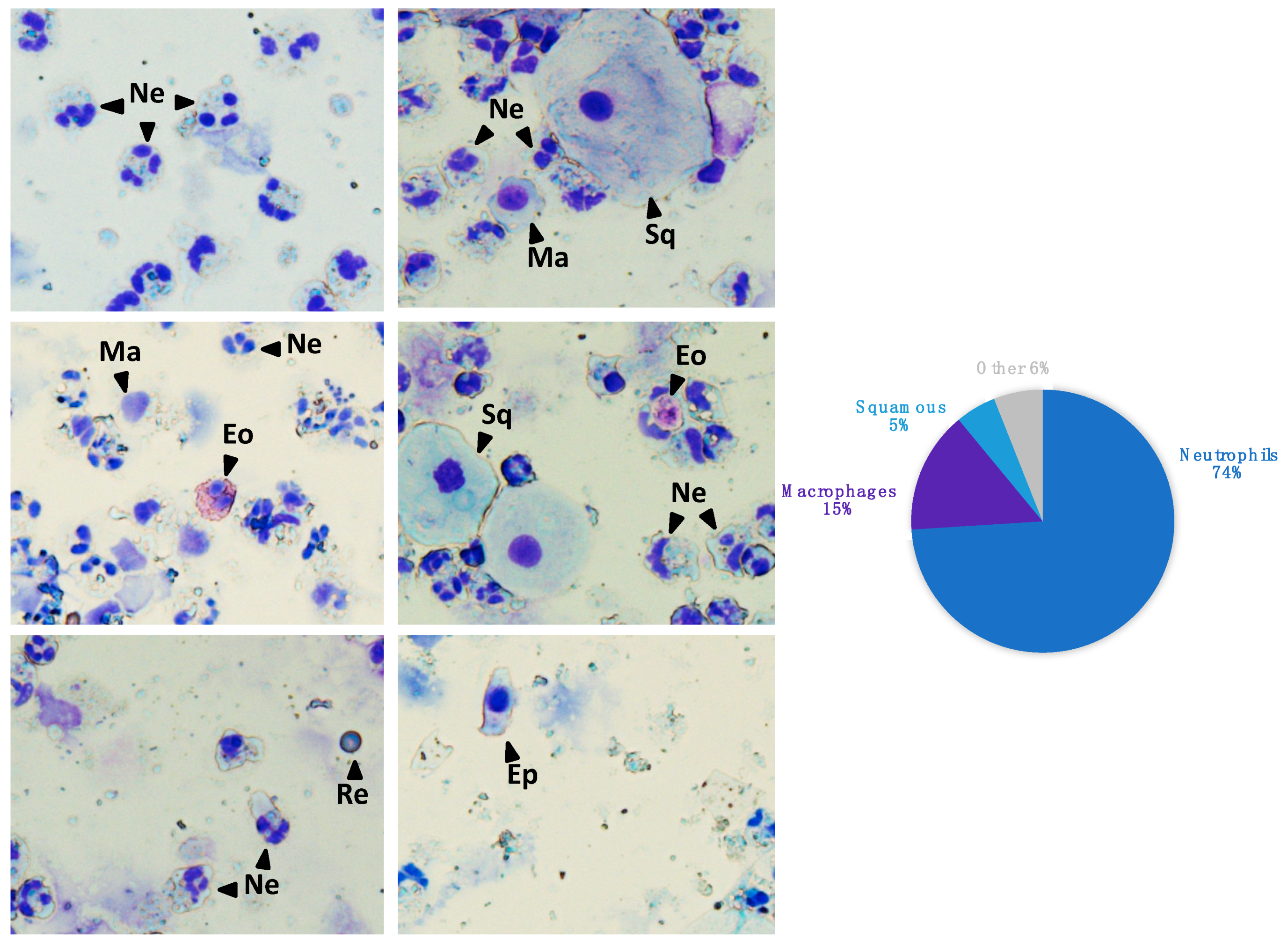
2.2. Sputum Samples Processing
2.3. Pyrosequencing DNA Methylation Analysis
2.4. Statistical Analyses
2.5. Enhancers Prediction
3. Results
3.1. MethylBiomark Cohort and Biobank
3.2. Biomarker Discovery in Nasal Epithelial Cell Samples from the MethylCF Cohort
3.3. Biomarker Assessment in Sputum Samples from the MethylBiomark Cohort
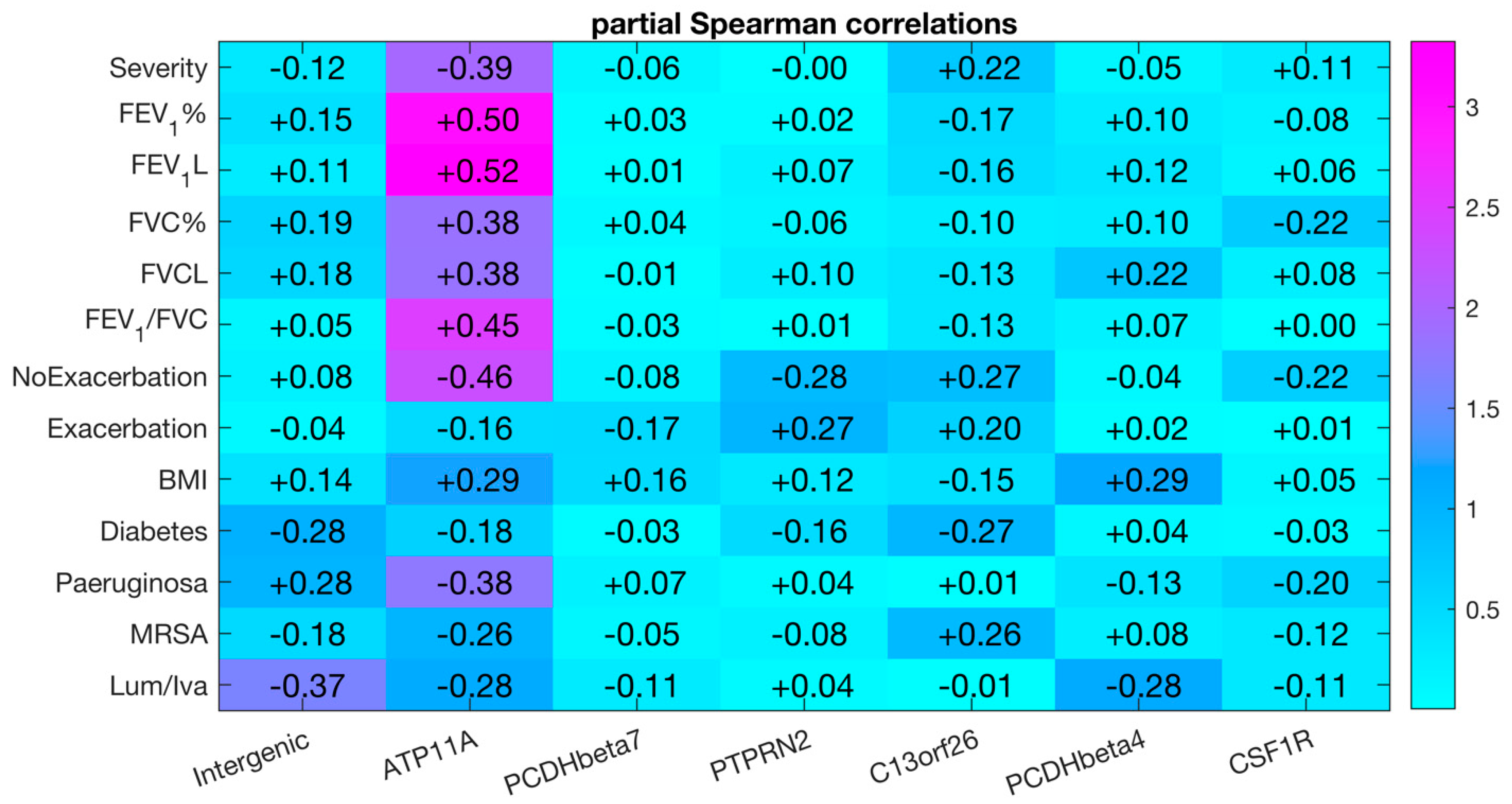
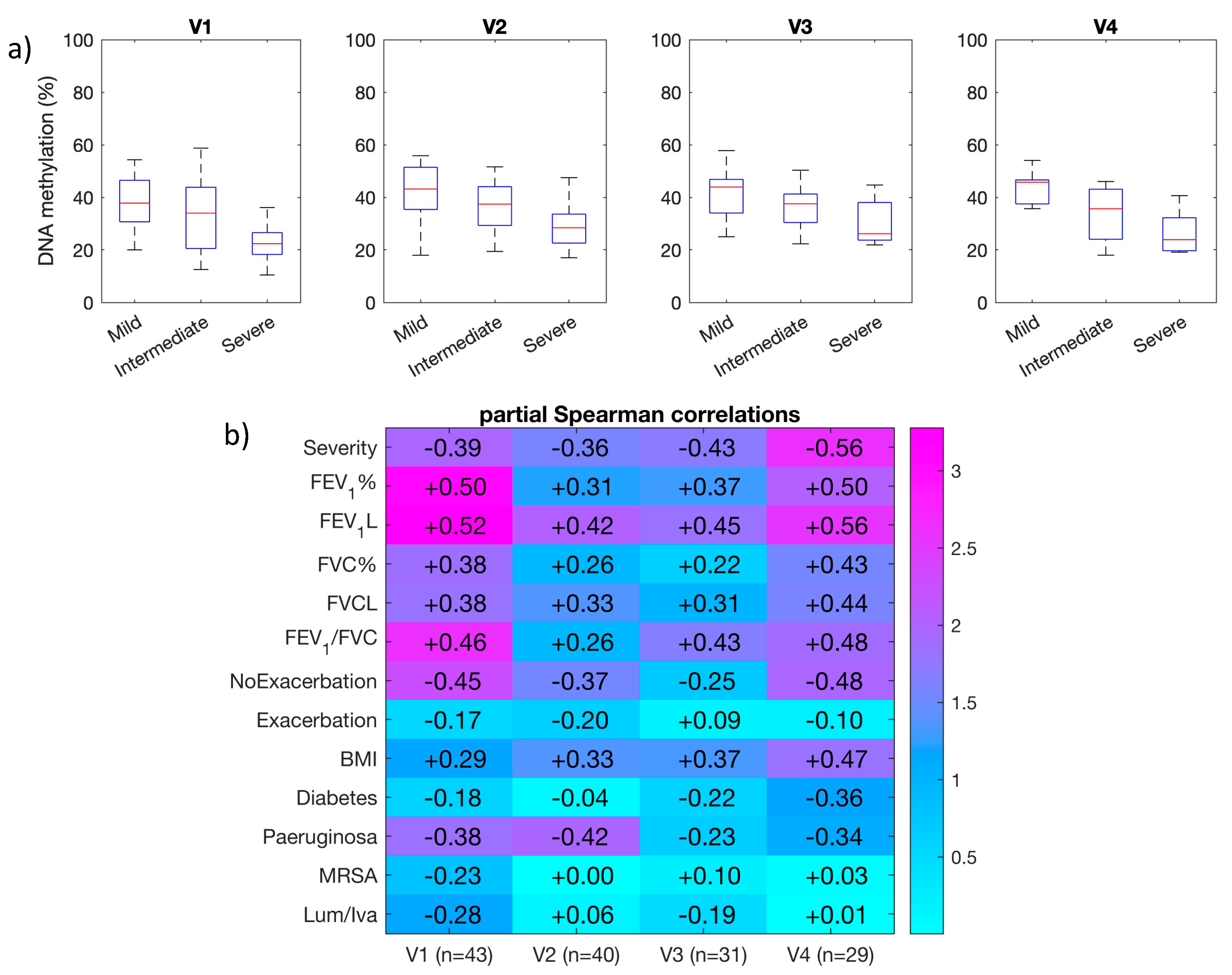
3.4. Longitudinal Analysis of DNA Methylation at ATP11A (cg11702988) in Sputum Samples
3.5. The cg11702988 (ATP11A) Maps to a Predicted Enhancer in the Lung
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat Rev Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Alton, E.W.; Bush, A. Cystic fibrosis. BMJ 2007, 335, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; van Koningsbruggen-Rietschel, S.; Nichols, D.P.; VanDevanter, D.R.; Davies, J.C.; Lee, T.; Durmowicz, A.G.; Ratjen, F.; Konstan, M.W.; Pearson, K.; et al. Building global development strategies for cf therapeutics during a transitional cftr modulator era. J. Cyst. Fibros. 2020, S1569-1993, 30161–30162. [Google Scholar] [CrossRef] [PubMed]
- Tiddens, H.A.; Puderbach, M.; Venegas, J.G.; Ratjen, F.; Donaldson, S.H.; Davis, S.D.; Rowe, S.M.; Sagel, S.D.; Higgins, M.; Waltz, D.A. Novel outcome measures for clinical trials in cystic fibrosis. Pediatr Pulmonol. 2015, 50, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Clancy, J.P.; Heltshe, S.L.; Ziady, A.; Kelley, T.; Accurso, F.; Pilewski, J.; Mayer-Hamblett, N.; Joseloff, E.; Sagel, S.D. Biomarkers for cystic fibrosis drug development. J. Cyst. Fibros. 2016, 15, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, S.C.; Parsons, F.; Gangell, C.; Brennan, S.; Stick, S.M.; Sly, P.D.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis. Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax 2011, 66, 408–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girón-Moreno, R.M.; Justicia, J.L.; Yamamoto, S.; Valenzuela, C.; Cisneros, C.; Gómez-Punter, R.M.; Fernandes-Vasconcelos, G.; Ancochea, J. Role of C-reactive protein as a biomarker for prediction of the severity of pulmonary exacerbations in patients with cystic fibrosis. BMC Pulm. Med. 2014, 14, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer-Hamblett, N.; Aitken, M.L.; Accurso, F.J.; Kronmal, R.A.; Konstan, M.W.; Burns, J.L.; Sagel, S.D.; Ramsey, B.W. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 822–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nick, J.A.; Sanders, L.A.; Ickes, B.; Briones, N.J.; Caceres, S.M.; Malcolm, K.C.; Brayshaw, S.J.; Chacon, C.S.; Barboa, C.M.; Jones, M.C.; et al. Blood mRNA biomarkers for detection of treatment response in acute pulmonary exacerbations of cystic fibrosis. Thorax 2013, 68, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quon, B.S.; Dai, D.L.; Hollander, Z.; Ng, R.T.; Tebbutt, S.J.; Man, S.F.; Wilcox, P.G.; Sin, D.D. Discovery of novel plasma protein biomarkers to predict imminent cystic fibrosis pulmonary exacerbations using multiple reaction monitoring mass spectrometry. Thorax 2016, 71, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, N.L.; Pereira, T.N.; Lewindon, P.J.; Shepherd, R.W.; Ramm, G.A. Circulating microRNAs as noninvasive diagnostic biomarkers of liver disease in children with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhães, M.; Tost, J.; Pineau, F.; Rivals, I.; Busato, F.; Alary, N.; Mely, L.; Leroy, S.; Murris, M.; Caimmi, D.; et al. Dynamic changes of DNA methylation and lung disease in cystic fibrosis: Lessons from a monogenic disease. Epigenomics 2018, 10, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Armstrong, D.A.; Salas, L.A.; Hazlett, H.F.; Nymon, A.B.; Dessaint, J.A.; Aridgides, D.S.; Mellinger, D.L.; Liu, X.; Christensen, B.C.; et al. Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin. Epigenet. 2018, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, M.; Rivals, I.; Claustres, M.; Varilh, J.; Thomasset, M.; Bergougnoux, A.; Mely, L.; Leroy, S.; Corvol, H.; Guillot, L.; et al. DNA methylation at modifier genes of lung disease severity is altered in cystic fibrosis. Clin. Epigenet. 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Pineau, F.; Caimmi, D.; Magalhães, M.; Fremy, E.; Mohamed, A.; Mely, L.; Leroy, S.; Murris, M.; Claustres, M.; Chiron, R.; et al. Blood co-expression modules identify potential modifier genes of diabetes and lung function in cystic fibrosis. PLoS ONE 2020, 15, e0231285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergougnoux, A.; Claustres, M.; De Sario, A. Nasal epithelial cells: A tool to study DNA methylation in airway diseases. Epigenomics 2015, 7, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schluchter, M.D.; Konstan, M.W.; Drumm, M.L.; Yankaskas, J.R.; et Knowles, M.R. Classifying severity of cystic fibrosis lung disease using longitudinal pulmonary function data. Am. J. Respir. Crit. Care Med. 2006, 174, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Gentzsch, M.; Mall, M.A. Ion Channel Modulators in Cystic Fibrosis. Chest 2018, 154, 383–393. [Google Scholar] [CrossRef] [PubMed]
- GTEx consortium. The genotype-tissue expression (GTEx) project. Nat Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein. Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Clews, J.; Ciuta, A.D.; Martin, E.R.; Ford, R.C. CFTR structure, stability, function and regulation. Biol. Chem. 2019, 400, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Bene, Z.; Fejes, Z.; Macek, M., Jr.; Amaral, M.D.; Balogh, I.; Nagy, B., Jr. Laboratory biomarkers for lung disease severity and progression in cystic fibrosis. Clin. Chim. Acta 2020, 508, 277–286, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Yanagihashi, Y.; Yamada, K.; Suzuki, C.; Uchiyama, Y.; Nagata, S. Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, 12212–12217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Mark, V.A.; Ghiboub, M.; Marsman, C.; Zhao, J.; van Dijk, R.; Hiralall, J.K.; Ho-Mok, K.S.; Castricum, Z.; de Jonge, W.J.; Oude Elferink, R.P.; et al. Phospholipid flippases attenuate LPS-induced TLR4 signaling by mediating endocytic retrieval of Toll-like receptor 4. Cell Mol. Life Sci. 2017, 74, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.; De Sario, A. DNA methylation changes in cystic fibrosis: Cause or consequence? Clin. Genet. 2020, 98, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Tom, R.G.; Hashem, A.S.; Gibran, H.; Josine, L.M.; Geoff, W.; Oliver, L.; Jie, Z.; Aparna, D.; McArdle, W.L.; Ho, K.; et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Wan, H.; Mei, S.; Ruan, H.; Zhang, Z.; Liu, C.; Guo, A.Y.; Diao, L.; Miao, X.; Han, L. Pancan-meQTL: A database to systematically evaluate the effects of genetic variants on methylation in human cancer. Nucleic Acids Res. 2019, 47, D1066–D1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Correlation with FEV1% | Differential DNA Methylation | ||||
|---|---|---|---|---|---|
| CpG | Gene | Coefficient | p-Value | ∆β (Mild-Severe) † | p-Value § |
| cg10582608 | Intergenic | 0.14 | 4.53 × 10−1 | 0.19 (0.34–0.15) | 1.9 × 10−3 |
| cg11702988 | ATP11A | 0.14 | 4.59 × 10−1 | 0.43 (0.78–0.35) | 3.2 × 10−3 |
| cg17735593 | PCDHβ7 | 0.20 | 2.77 × 10−1 | 0.17 (0.66–0.49) | 5.3 × 10−3 |
| cg23299919 | PTPRN2 | 0.45 | 1.23 × 10−2 | 0.25 (0.45–0.20) | 5.6 × 10−4 |
| cg08379987 | C13orf26 | 0.67 | 5.70 × 10−5 | 0.19 (0.57–0.38) | 3.2 × 10−3 |
| cg06048354 | PCDHβ4 | 0.39 | 3.16 × 10−2 | 0.16 (0.41–0.25) | 6.7 × 10−3 |
| cg05524038 | CSF1R | 0.49 | 5.83 × 10−3 | 0.13 (0.79–0.66) | 1.2 × 10−4 |
| V1 | V2 | V3 | V4 | |
|---|---|---|---|---|
| Age, year | 21.5 (7.8) | 21.7 (9.4) | 22.4 (8.6) | 22.5 (10.2) |
| Sex ratio (M:F) | 26:17 | 22:18 | 18:14 | 16:13 |
| CFTR genotype | ||||
| p.Phe508del-p.Phe508del | 34 | 30 | 25 | 19 |
| p.Phe508del-other variant ¶ | 9 | 9 | 7 | 9 |
| p.Arg553*-p.Trp1282* | 0 | 1 | 0 | 1 |
| Mild | 8 | 9 | 8 | 6 |
| Intermediate | 26 | 22 | 16 | 15 |
| Severe | 9 | 9 | 8 | 8 |
| Weight, kg | 57.0 (15.0) | 56.0 (15.5) | 58.0 (17.5) | 54.0 (11.8) |
| Height, cm | 166.0 (15.0) | 167.0 (14.5) | 169 (14.5) | 167 (12.5) |
| Body mass index, kg/m2 | 19.9 (3.0) | 19.7 (2.9) | 20.3 (3.4) | 19.8 (2.1) |
| FEV1,% predicted | 63.5 (31.5) | 66.0 (36.7) | 65.0 (41.5) | 59.0 (40.8) |
| FEV1, liters | 2.2 (1.0) | 2.1 (1.5) | 2.1 (1.5) | 2.0 (1.5) |
| FVC,% predicted | 81.0 (21.5) | 79.0 (27.5) | 83.0 (27.0) | 82.0 (27.5) |
| FVC, liters | 3.4 (1.0) | 3.2 (1.2) | 3.4 (1.6) | 3.1 (1.6) |
| Exacerbation # | 12% | 35% | 35% | 60% |
| Pancreatic insufficiency | 100% | 100% | 100% | 100% |
| Diabetes † | 12% | 20% | 19% | 20% |
| Atopy | 35% | 40% | 42% | 30% |
| Pseudomonas aeruginosa‡ | 72% | 65% | 61% | 60% |
| Aspergillus fumigatus‡ | 26% | 25% | 23% | 20% |
| MRSA ‡ | 16% | 22% | 19% | 20% |
| Other germs § | 81% | 75% | 77% | 70% |
| Azithromycin | 79% | 72% | 68% | 80% |
| Tobramycine | 30% | 22% | 16% | 20% |
| Colistin | 58% | 55% | 55% | 60% |
| Aztreonam | 19% | 20% | 19% | 10% |
| Nasal corticosteroid | 30% | 37% | 35% | 40% |
| lumacaftor/ivacaftor | 47% | 42% | 45% | 30% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pineau, F.; Caimmi, D.; Taviaux, S.; Reveil, M.; Brosseau, L.; Rivals, I.; Drevait, M.; Vachier, I.; Claustres, M.; Chiron, R.; et al. DNA Methylation at ATP11A cg11702988 Is a Biomarker of Lung Disease Severity in Cystic Fibrosis: A Longitudinal Study. Genes 2021, 12, 441. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030441
Pineau F, Caimmi D, Taviaux S, Reveil M, Brosseau L, Rivals I, Drevait M, Vachier I, Claustres M, Chiron R, et al. DNA Methylation at ATP11A cg11702988 Is a Biomarker of Lung Disease Severity in Cystic Fibrosis: A Longitudinal Study. Genes. 2021; 12(3):441. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030441
Chicago/Turabian StylePineau, Fanny, Davide Caimmi, Sylvie Taviaux, Maurane Reveil, Laura Brosseau, Isabelle Rivals, Margot Drevait, Isabelle Vachier, Mireille Claustres, Raphaël Chiron, and et al. 2021. "DNA Methylation at ATP11A cg11702988 Is a Biomarker of Lung Disease Severity in Cystic Fibrosis: A Longitudinal Study" Genes 12, no. 3: 441. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030441