
A Missense Mutation in the KLF7 Gene Is a Potential Candidate Variant for Congenital Deafness in Australian Stumpy Tail Cattle Dogs

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Phenotyping and Samples
2.3. Next Generation Sequencing and Variant Calling
2.4. Genome Wide Association Analysis
2.5. Next Generation Sequencing Data Analysis for Identification of Associated Variants
2.6. Genotyping of KLF7 Variant in Australian Stumpy Tail Cattle Dogs
2.7. Investigation of Human Deafness Genes in 3 Deaf Australian Stumpy Tail Cattle Dogs
3. Results
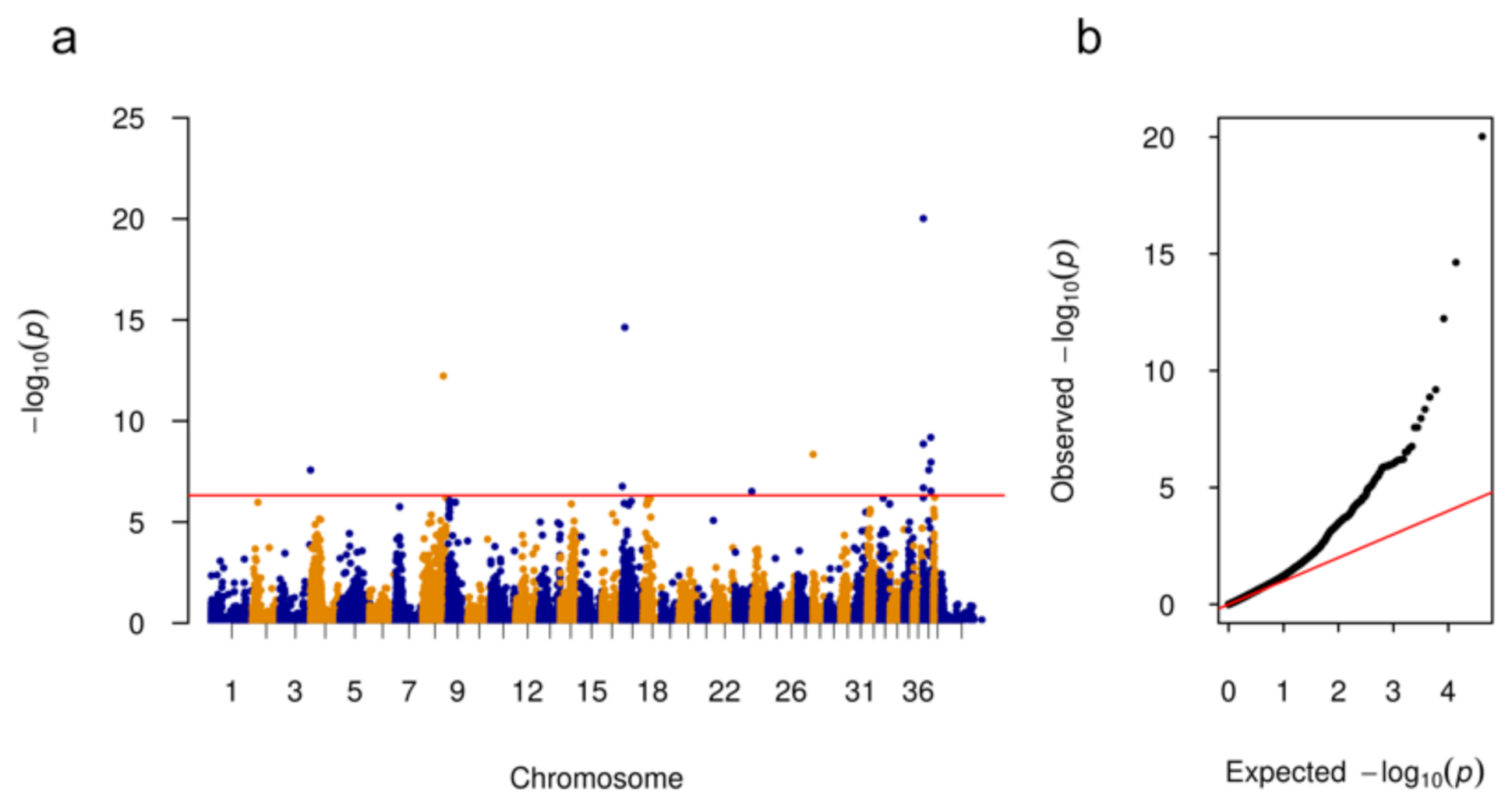
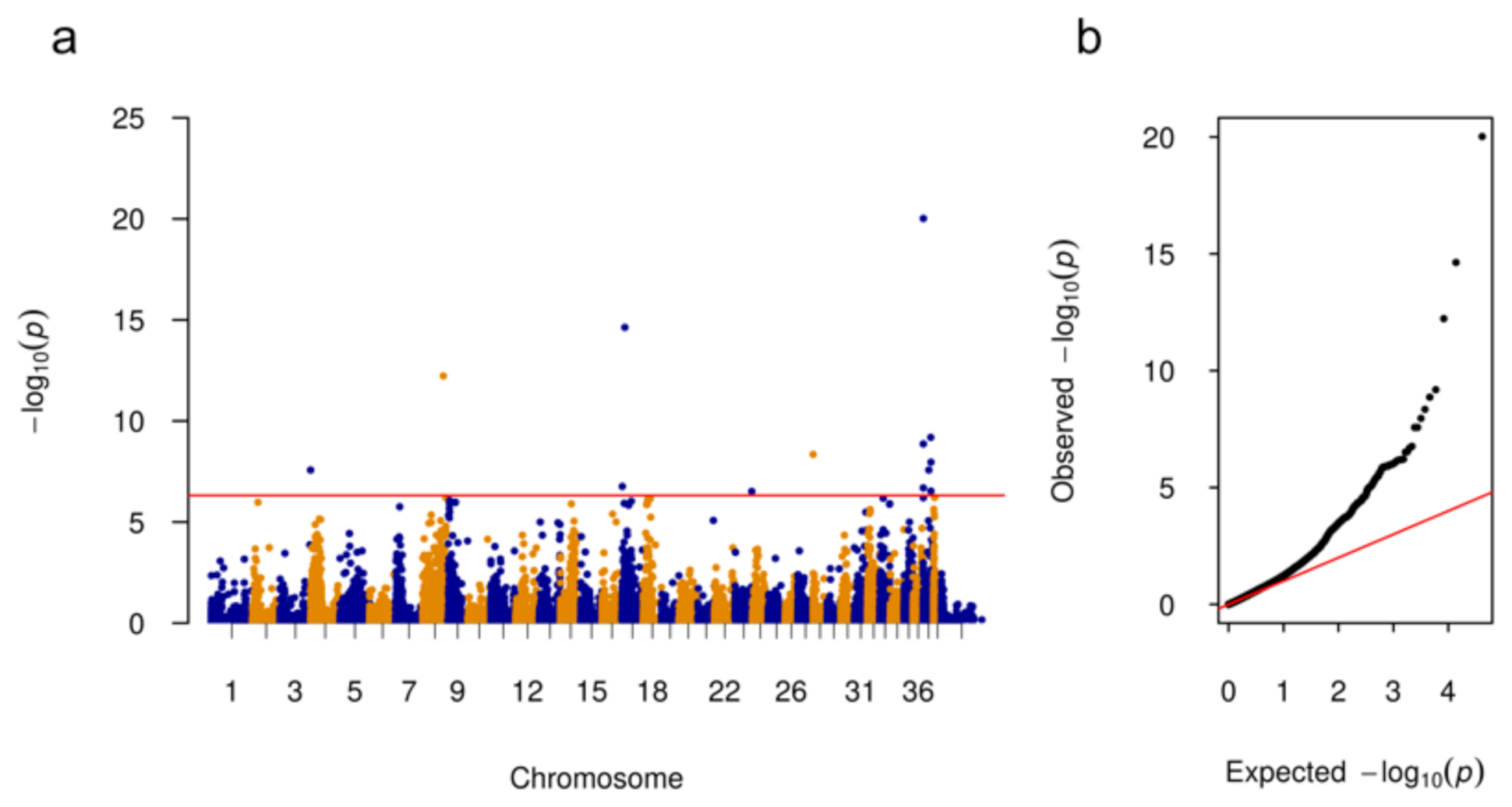
3.1. Genome Wide Association Analysis
3.2. Whole Genome Sequencing Reveals Four Potential Variants
3.3. Genotyping of KLF7 Variant in ASCDs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rak, S.G.; Distl, O. Congenital sensorineural deafness in dogs: A molecular genetic approach toward unravelling the responsible genes. Vet. J. 2005, 169, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Smith, M.; Strain, G.M. STRING Data Mining of GWAS Data in Canine Hereditary Pigment-Associated Deafness. Vet. Anim. Sci. 2020, 9, 100118. [Google Scholar] [CrossRef] [PubMed]
- Strain, G.M. Deafness prevalence and pigmentation and gender associations in dog breeds at risk. Vet. J. 2004, 167, 23–32. [Google Scholar] [CrossRef]
- Strain, G.M. The genetics of deafness in domestic animals. Front. Vet. Sci. 2015, 2, 29. [Google Scholar] [CrossRef]
- Strain, G.M. Aetiology, prevalence and diagnosis of deafness in dogs and cats. Br. Vet. J. 1996, 152, 17–36. [Google Scholar] [CrossRef]
- Cunningham, L.L.; Tucci, D.L. Hearing loss in adults. N. Engl. J. Med. 2017, 377, 2465–2473. [Google Scholar] [CrossRef]
- Hiraide, F.; Paparella, M.M. Histopathology of the temporal bones of deaf dogs. Auris Nasus Larynx 1988, 15, 97–104. [Google Scholar] [CrossRef]
- Coppens, A.; Resibois, A.; Poncelet, L. Bilateral deafness in a maltese terrier and a great pyrenean puppy: Inner ear morphology. J. Comp. Pathol. 2000, 122, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Strain, G.M. Hereditary deafness in dogs and cats: Causes, prevalence, and current research. Strain 2003, 225, 578–9758. [Google Scholar]
- Sims, M.H. Electrodiagnostic evaluation of auditory function. Vet. Clin. N. Am. Small Anim. Pract. 1988, 18, 913–944. [Google Scholar] [CrossRef]
- Wilson, W.J.; Mills, P.C. Brainstem auditory-evoked response in dogs. Am. J. Vet. Res. 2005, 66, 2177–2187. [Google Scholar] [CrossRef]
- Sommerlad, S.; McRae, A.F.; McDonald, B.; Johnstone, I.; Cuttell, L.; Seddon, J.M.; O’Leary, C.A. Congenital sensorineural deafness in Australian stumpy-tail cattle dogs is an autosomal recessive trait that maps to CFA10. PLoS ONE 2010, 5, e13364. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, J.S.; Lam, E.T.; Ruhe, A.L.; Erdman, C.A.; Robertson, K.R.; Webb, A.A.; Williams, D.C.; Chang, M.L.; Hytönen, M.K.; Lohi, H. Variation in genes related to cochlear biology is strongly associated with adult-onset deafness in border collies. PLoS Genet. 2012, 8, e13364. [Google Scholar] [CrossRef]
- Guevar, J.; Olby, N.J.; Meurs, K.M.; Yost, O.; Friedenberg, S.G. Deafness and vestibular dysfunction in a Doberman Pinscher puppy associated with a mutation in the PTPRQ gene. J. Vet. Intern. Med. 2018, 32, 665–669. [Google Scholar] [CrossRef]
- Webb, A.A.; Ruhe, A.L.; Neff, M.W. A missense mutation in MYO7A is associated with bilateral deafness and vestibular dysfunction in the Doberman pinscher breed. Can. J. Vet. Res. 2019, 83, 142–148. [Google Scholar]
- Kluth, S.; Distl, O. Congenital sensorineural deafness in Dalmatian dogs associated with quantitative trait loci. PLoS ONE 2013, 8, e1002898. [Google Scholar] [CrossRef]
- Hayward, J.J.; Kelly-Smith, M.; Boyko, A.R.; Burmeister, L.; De Risio, L.; Mellersh, C.; Freeman, J.; Strain, G.M. A genome-wide association study of deafness in three canine breeds. PLoS ONE 2020, 15, e0232900. [Google Scholar] [CrossRef]
- Laub, F.; Lei, L.; Sumiyoshi, H.; Kajimura, D.; Dragomir, C.; Smaldone, S.; Puche, A.C.; Petros, T.J.; Mason, C.; Parada, L.F. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol. Cell. Biol. 2005, 25, 5699–5711. [Google Scholar] [CrossRef]
- Chen, J.; Tambalo, M.; Barembaum, M.; Ranganathan, R.; Simões-Costa, M.; Bronner, M.E.; Streit, A. A systems-level approach reveals new gene regulatory modules in the developing ear. Development 2017, 144, 1531–1543. [Google Scholar] [CrossRef]
- Strain, G. Brainstem auditory evoked response (BAER). Deaf. Dogs Cats 2011, 83–107. [Google Scholar] [CrossRef]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Consortium, D.B.V.D.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using QQ and manhattan plots. Biorxiv 2014, 005165. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Clarke, G.M.; Anderson, C.A.; Pettersson, F.H.; Cardon, L.R.; Morris, A.P.; Zondervan, K.T. Basic statistical analysis in genetic case-control studies. Nat. Protoc. 2011, 6, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Jourquin, J.; Duncan, D.; Shi, Z.; Zhang, B. GLAD4U: Deriving and prioritizing gene lists from PubMed literature. BMC Genom. 2012, 13, S20. [Google Scholar] [CrossRef]
- Taylor, W. Residual colours: A proposal for aminochromography. Protein Eng. 1997, 10, 743–746. [Google Scholar] [CrossRef]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.; Freeman, J.; De Risio, L. Decline in prevalence of congenital sensorineural deafness in Dalmatian dogs in the United Kingdom. J. Vet. Intern. Med. 2020, 34, 1524–1531. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; Van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858. [Google Scholar] [CrossRef]
- Raven, L.-A.; Cocks, B.G.; Hayes, B.J. Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genom. 2014, 15, 62. [Google Scholar] [CrossRef]
- Yan, J.; Wang, X. Detection of Disease-associated Mutations and Biomarkers Using Next-generation Sequencing. Detect. Methods Precis. Med. 2020, 18, 119. [Google Scholar]
- Donat, S.; Lourenço, M.; Paolini, A.; Otten, C.; Renz, M.; Abdelilah-Seyfried, S. Heg1 and Ccm1/2 proteins control endocardial mechanosensitivity during zebrafish valvulogenesis. Elife 2018, 7, e28939. [Google Scholar] [CrossRef] [PubMed]
- Laub, F.; Aldabe, R.; Friedrich Jr, V.; Ohnishi, S.; Yoshida, T.; Ramirez, F. Developmental expression of mouse Krüppel-like transcription factor KLF7 suggests a potential role in neurogenesis. Dev. Biol. 2001, 233, 305–318. [Google Scholar] [CrossRef]
- Blackmore, M.G.; Wang, Z.; Lerch, J.K.; Motti, D.; Zhang, Y.P.; Shields, C.B.; Lee, J.K.; Goldberg, J.L.; Lemmon, V.P.; Bixby, J.L. Krüppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc. Natl. Acad. Sci. USA 2012, 109, 7517–7522. [Google Scholar] [CrossRef]
- Lei, L.; Laub, F.; Lush, M.; Romero, M.; Zhou, J.; Luikart, B.; Klesse, L.; Ramirez, F.; Parada, L.F. The zinc finger transcription factor Klf7 is required for TrkA gene expression and development of nociceptive sensory neurons. Genes Dev. 2005, 19, 1354–1364. [Google Scholar] [CrossRef]
- Kajimura, D.; Dragomir, C.; Ramirez, F.; Laub, F. Identification of genes regulated by transcription factor KLF7 in differentiating olfactory sensory neurons. Gene 2007, 388, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Akçimen, F.; Vural, A.; Durmuş, H.; Çakar, A.; Houlden, H.; Parman, Y.G.; Başak, A.N. A novel homozygous FBXO38 variant causes an early-onset distal hereditary motor neuronopathy type IID. J. Hum. Genet. 2019, 64, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Tambalo, M.; Anwar, M.; Ahmed, M.; Streit, A. Enhancer activation by FGF signalling during otic induction. Dev. Biol. 2020, 457, 69–82. [Google Scholar] [CrossRef]
- Brophy, P.D.; Alasti, F.; Darbro, B.W.; Clarke, J.; Nishimura, C.; Cobb, B.; Smith, R.J.; Manak, J.R. Genome-wide copy number variation analysis of a Branchio-oto-renal syndrome cohort identifies a recombination hotspot and implicates new candidate genes. Hum. Genet. 2013, 132, 1339–1350. [Google Scholar] [CrossRef]
- Wells, H.R.; Freidin, M.B.; Abidin, F.N.Z.; Payton, A.; Dawes, P.; Munro, K.J.; Morton, C.C.; Moore, D.R.; Dawson, S.J.; Williams, F.M. GWAS Identifies 44 Independent Associated Genomic Loci for Self-Reported Adult Hearing Difficulty in UK Biobank. Am. J. Hum. Genet. 2019, 105, 788–802. [Google Scholar] [CrossRef]
- Kalra, G.; Milon, B.; Casella, A.M.; Herb, B.R.; Humphries, E.; Song, Y.; Rose, K.P.; Hertzano, R.; Ament, S.A. Biological insights from multi-omic analysis of 31 genomic risk loci for adult hearing difficulty. PLoS Genet. 2020, 16, e1009025. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, Z.; Zhang, X.; Cui, T.; Zhang, W.; Shao, S.; Li, H.; Wang, N. GATA binding protein 3 is a direct target of Kruppel-like transcription factor 7 and inhibits chicken adipogenesis. Front. Physiol. 2020, 11, 610. [Google Scholar] [CrossRef]
- Lawoko-Kerali, G.; Rivolta, M.N.; Holley, M. Expression of the transcription factors GATA3 and Pax2 during development of the mammalian inner ear. J. Comp. Neurol. 2002, 442, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Groenen, P.; Nesbit, M.A.; Schuffenhauer, S.; Lichtner, P.; Vanderlinden, G.; Harding, B.; Beetz, R.; Bilous, R.W.; Holdaway, I. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 2000, 406, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Pierrat, M.-J.; Marsaud, V.; Mauviel, A.; Javelaud, D. Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-β. J. Biol. Chem. 2012, 287, 17996–18004. [Google Scholar] [CrossRef]
- Karlsson, E.K.; Baranowska, I.; Wade, C.M.; Hillbertz, N.H.S.; Zody, M.C.; Anderson, N.; Biagi, T.M.; Patterson, N.; Pielberg, G.R.; Kulbokas, E.J. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat. Genet. 2007, 39, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, W.; Ren, L.; Yang, M.; Zhao, Y.; Guo, Z.; Yi, H.; Li, M.; Hu, Y.; Long, X. A de novo silencer causes elimination of MITF-M expression and profound hearing loss in pigs. BMC Biol. 2016, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Philipp, U.; Lupp, B.; Mömke, S.; Stein, V.; Tipold, A.; Eule, J.C.; Rehage, J.; Distl, O. A MITF mutation associated with a dominant white phenotype and bilateral deafness in German Fleckvieh cattle. PLoS ONE 2011, 6, e28857. [Google Scholar] [CrossRef]
- Tassabehji, M.; Newton, V.E.; Read, A.P. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 1994, 8, 251–255. [Google Scholar] [CrossRef]
- Strain, G.M. Canine deafness. Vet. Clin. Small Anim. Pract. 2012, 42, 1209–1224. [Google Scholar] [CrossRef]
- De Risio, L.; Freeman, J.; Lewis, T. Prevalence, heritability and genetic correlations of congenital sensorineural deafness and coat pigmentation phenotype in the English bull terrier. BMC Vet. Res. 2016, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Sommerlad, S.F.; Morton, J.M.; Haile-Mariam, M.; Johnstone, I.; Seddon, J.M.; O’Leary, C.A. Prevalence of congenital hereditary sensorineural deafness in Australian Cattle Dogs and associations with coat characteristics and sex. BMC Vet. Res. 2012, 8, 202. [Google Scholar] [CrossRef]
- Strain, G.M.; Kearney, M.T.; Gignac, I.J.; Levesque, D.C.; Nelson, H.J.; Tedford, B.L.; Remsen, L.G. Brainstem auditory-evoked potential assessment of congenital deafness in Dalmatians: Associations with phenotypic markers. J. Vet. Intern. Med. 1992, 6, 175–182. [Google Scholar] [CrossRef]
- Greibrokk, T. Hereditary deafness in the Dalmation: Relationship to eye and coat color. Journal 1995, 30, 170–176. [Google Scholar]
- Famula, T.; Oberbauer, A.; Sousa, C. A threshold model analysis of deafness in Dalmatians. Mamm. Genome 1996, 7, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.; Lakhani, K. Prevalence and prevention of deafness in the Dalmatian—Assessing the effect of parental hearing status and gender using ordinary logistic and generalized random litter effect models. Vet. J. 1997, 154, 121–133. [Google Scholar] [CrossRef]
- Muhle, A.C.; Jaggy, A.; Stricker, C.; Steffen, F.; Dolf, G.; Busato, A.; Kornberg, M.; Mariscoli, M.; Srenk, P.; Gaillard, C. Further contributions to the genetic aspect of congenital sensorineural deafness in Dalmatians. Vet. J. 2002, 163, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Juraschko, K.; Meyer-Lindenberg, A.; Nolte, I.; Distl, O. Analysis of systematic effects on congenital sensorineural deafness in German Dalmatian dogs. Vet. J. 2003, 166, 164–169. [Google Scholar] [CrossRef]
- Cargill, E.; Famula, T.; Strain, G.; Murphy, K. Heritability and segregation analysis of deafness in US Dalmatians. Genetics 2004, 166, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Metallinos, D.; Rine, J. Exclusion of EDNRB and KIT as the basis for white spotting in Border Collies. Genome Biol. 2000, 1, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Stritzel, S.; Wohlke, A.; Distl, O. Elimination of SILV as a candidate for congenital sensorineural deafness in Dalmatian dogs. Anim. Genet. 2007, 38, 662. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| ID | Gender | Coat Colour | BAER Test Results |
|---|---|---|---|
| 217 | Female | Red speckled | Bilaterally Deaf |
| 253 | Female | Red speckled | Bilaterally Deaf |
| 330 | Female | Red | Bilaterally Deaf |
| 326 | Female | Red speckled | Normal Hearing |
| CFA | Position | p-Value | Nearby Genes | Distance (bp) |
|---|---|---|---|---|
| 3 | 90,987,932 | 2.67 × 10−8 | LCORL | 186,575 |
| 8 | 62,032,863 | 5.93 × 10−13 | DGLUCY | 19,884 |
| 17 | 1,977,343 | 1.73 × 10−7 | EIPR1 | 0 |
| 17 | 9,456,133 | 2.34 × 10−15 | TRIB2 | 204,307 |
| 23 | 50,096,314 | 3.04 × 10−7 | KCNAB1 | 0 |
| 28 | 21,516 | 4.50 × 10−9 | PTPN20 | 42,882 |
| 37 | 13,393 | 2.04 × 10−7 | WDR75 | 144,007 |
| 37 | 44,793 | 9.54 × 10−21 | WDR75 | 112,607 |
| 37 | 80,438 | 1.36 × 10−9 | WDR75 | 76,962 |
| 37 | 16,399,127 | 2.66 × 10−8 | CRYGD | 25,757 |
| 37 | 22,102,392 | 6.48 × 10−10 | ABCA12 | 34,340 |
| 37 | 22,579,983 | 2.93 × 10−7 | FN1 | 57,573 |
| 37 | 22,711,697 | 1.10 × 10−8 | FN1 | 189,287 |
| Chr | HGVS Genome Position (a) | Variant Type | Gene (b) | #217 | #253 | #330 | #326 |
|---|---|---|---|---|---|---|---|
| 13 | NC_006595.3:g.60805542 C>T | missense variant | GC | C_T | C_T | C_T | C_C |
| 21 | NC_006603.3:g.23019999 C>T | missense variant | MAP6 | C_T | C_T | C_T | C_C |
| 33 | NC_006615.3:g.28028412 G>C | missense variant | HEG1 | G_C | G_C | G_C | G_G |
| 37 | NC_006619.3:g.15562684 G>A | missense variant | KLF7 | A_A | A_A | A_G | G_G |
| Gene | Amino Acid Exchange | SIFT | Polyphen-2 | PROVEAN |
|---|---|---|---|---|
| GC | p.Gly389Rrg | Tolerated | Benign | Neutral |
| MAP6 | p.Arg486Cys | Affect protein function | Benign | Neutral |
| HEG1 | p.His531Asp | Affect protein function | Unknown | Deleterious |
| KLF7 | p.Leu173Phe | Affect protein function | Possibly damaging | Neutral |
| Phenotype | G_G | A_G | A_A | Total Number | P(c) |
|---|---|---|---|---|---|
| Unilaterally deaf | 10 | 10 | 1 | 21 | 0.054 |
| Bilaterally deaf | 3 | 4 | 3 | 10 | 0.010 |
| Deafness (uni (a) + bi (b)) | 13 | 14 | 4 | 31 | 0.014 |
| Normal hearing | 22 | 5 | 1 | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Shan, S.; Sommerlad, S.; Seddon, J.M.; Brenig, B. A Missense Mutation in the KLF7 Gene Is a Potential Candidate Variant for Congenital Deafness in Australian Stumpy Tail Cattle Dogs. Genes 2021, 12, 467. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040467
Xu F, Shan S, Sommerlad S, Seddon JM, Brenig B. A Missense Mutation in the KLF7 Gene Is a Potential Candidate Variant for Congenital Deafness in Australian Stumpy Tail Cattle Dogs. Genes. 2021; 12(4):467. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040467
Chicago/Turabian StyleXu, Fangzheng, Shuwen Shan, Susan Sommerlad, Jennifer M. Seddon, and Bertram Brenig. 2021. "A Missense Mutation in the KLF7 Gene Is a Potential Candidate Variant for Congenital Deafness in Australian Stumpy Tail Cattle Dogs" Genes 12, no. 4: 467. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040467