
Novel Compound Heterozygous Mutation in TRAPPC9 Gene: The Relevance of Whole Genome Sequencing

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
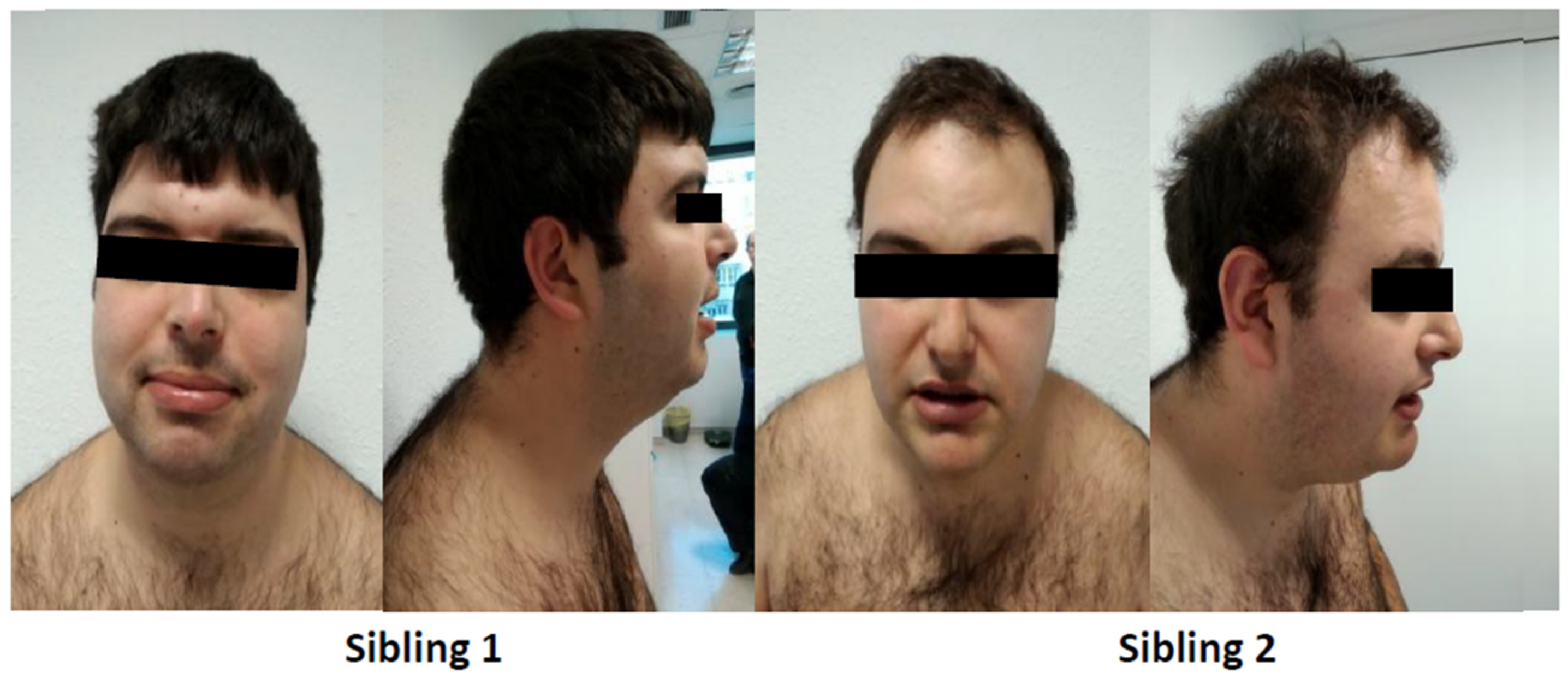
2.1. Case Report
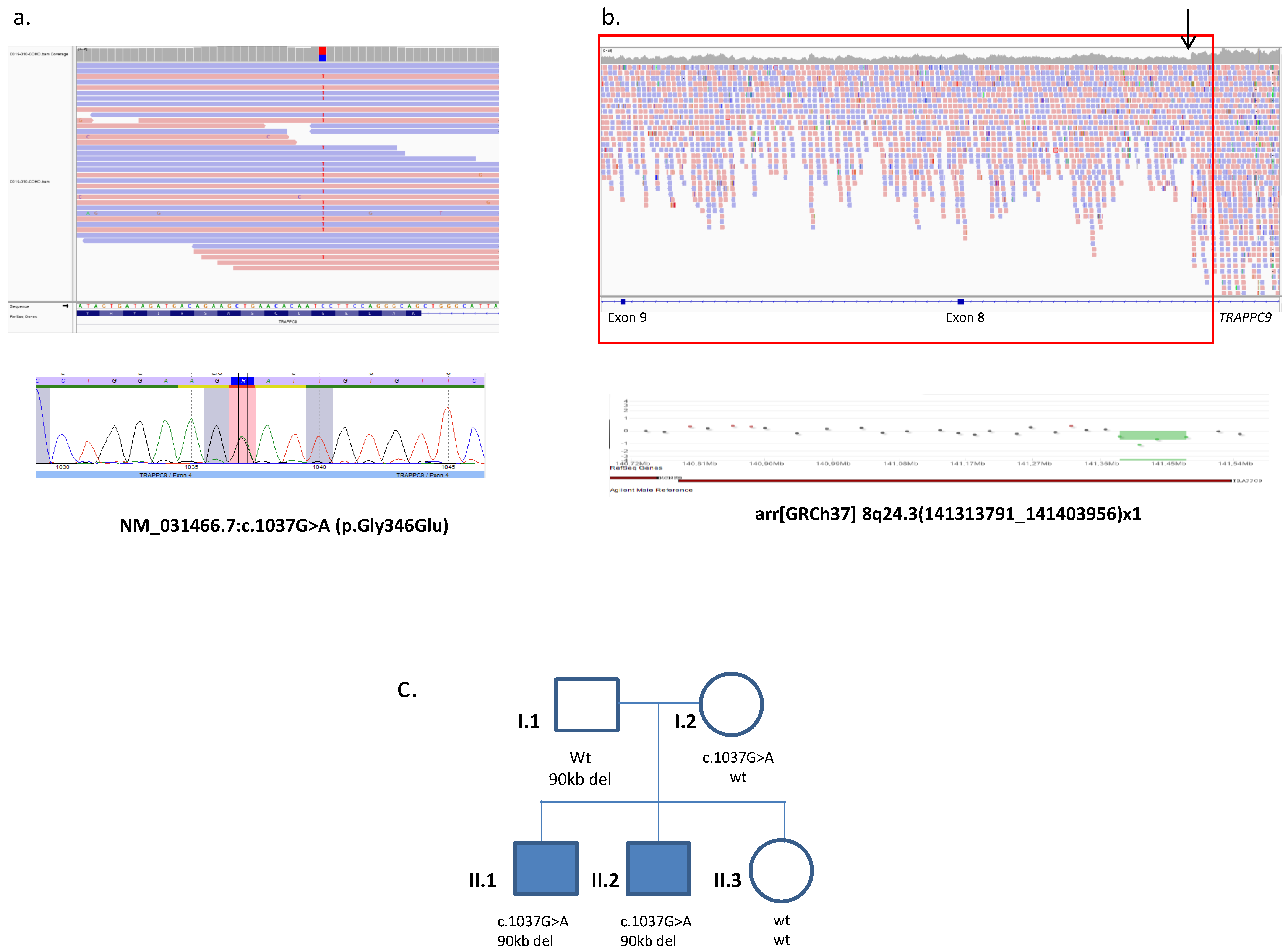
2.2. Whole Genome Sequencing and Data Analysis
2.3. Segregation Studies
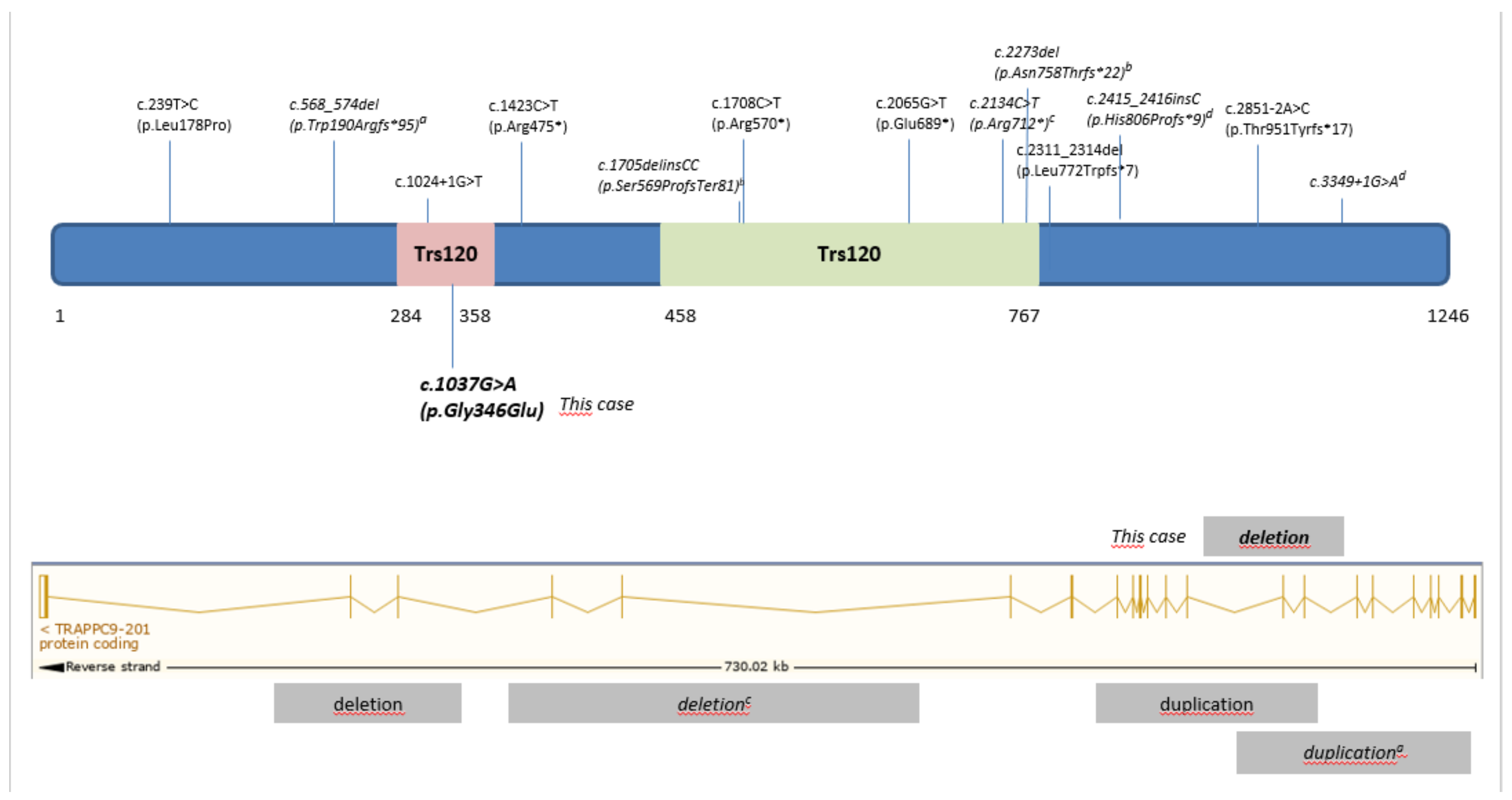
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finucane, B.M.; Ledbetter, D.H.; Vorstman, J.A. Diagnostic genetic testing for neurodevelopmental psychiatric disorders: Closing the gap between recommendation and clinical implementation. Curr. Opin. Genet. Dev. 2021, 68, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Charlson, F.J.; Norman, R.E.; Flaxman, A.D.; Johns, N.; et al. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1575–1586. [Google Scholar] [CrossRef]
- Polyak, A.; Rosenfeld, J.A.; Girirajan, S. An assessment of sex bias in neurodevelopmental disorders. Genome Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.; Girirajan, S. Mapping a shared genetic basis for neurodevelopmental disorders. Genome Med. 2017, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- Willsey, A.J.; Morris, M.T.; Wang, S.; Willsey, H.R.; Sun, N.; Teerikorpi, N.; Baum, T.B.; Cagney, G.; Bender, K.J.; Desai, T.A.; et al. The Psychiatric Cell Map Initiative: A Convergent Systems Biological Approach to Illuminating Key Molecular Pathways in Neuropsychiatric Disorders. Cell 2018, 174, 505–520. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- D’Haene, E.; Vergult, S. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet. Med. 2021, 23, 34–46. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Madrigal, I.; Rabionet, R.; Alvarez-Mora, M.I.; Sanchez, A.; Rodríguez-Revenga, L.; Estivill, X.; Mila, M. Spectrum of clinical heterogeneity of β-tubulin TUBB5 gene mutations. Gene 2019, 695, 12–17. [Google Scholar] [CrossRef]
- Mortreux, J.; Busa, T.; Germain, D.P.; Nadeau, G.; Puechberty, J.; Coubes, C.; Gatinois, V.; Cacciagli, P.; Duffourd, Y.; Pinard, J.M.; et al. The role of CNVs in the etiology of rare autosomal recessive disorders: The example of TRAPPC9-associated intellectual disability. Eur. J. Hum. Genet. 2018, 26, 143–148. [Google Scholar] [CrossRef]
- Sacher, M.; Shahrzad, N.; Kamel, H.; Milev, M.P. TRAPPopathies: An emerging set of disorders linked to variations in the genes encoding transport protein particle (TRAPP)-associated proteins. Traffic 2019, 20, 5–26. [Google Scholar] [CrossRef] [Green Version]
- Lipatova, Z.; Van Bergen, N.; Stanga, D.; Sacher, M.; Christodoulou, J.; Segev, N. TRAPPing a neurological disorder: From yeast to humans. Autophagy 2020, 16, 965–966. [Google Scholar] [CrossRef]
- Mbimba, T.; Hussein, N.J.; Najeed, A.; Safadi, F.F. TRAPPC9: Novel insights into its trafficking and signaling pathways in health and disease. Int. J. Mol. Med. 2018, 42, 2991–2997. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, B.; DeGruttola, A.; Zhu, Y.; Lin, Y.; Zhang, Y.; Mo, X.; Hu, W. Emerging role of NIK/IKK2-binding protein (NIBP)/trafficking protein particle complex 9 (TRAPPC9) in nervous system diseases. Transl. Res. 2020, 224, 55–70. [Google Scholar] [CrossRef]
- Zhang, Y.; Bitner, D.; Pontes Filho, A.A.; Li, F.; Liu, S.; Wang, H.; Yang, F.; Adhikari, S.; Gordon, J.; Srinivasan, S.; et al. Expression and function of NIK- and IKK2-binding protein (NIBP) in mouse enteric nervous system. Neurogastroenterol. Motil. 2014, 26, 77–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.; Weng, M.; Chhetri, G.; Usman, M.; Li, Y.; Yu, Q.; Ding, Y.; Wang, Z.; Wang, X.; Sultana, P.; et al. Trappc9 deficiency in mice impairs learning and memory by causing imbalance of dopamine D1 and D2 neurons. Sci. Adv. 2020, 6, eabb7781. [Google Scholar] [CrossRef] [PubMed]
- Hnoonual, A.; Graidist, P.; Kritsaneepaiboon, S.; Limprasert, P. Novel Compound Heterozygous Mutations in the TRAPPC9 Gene in Two Siblings with Autism and Intellectual Disability. Front. Genet. 2019, 10, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Z.; Kong, X. Diagnosis of a case with mental retardation due to novel compound heterozygous variants of TRAPPC9 gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2019, 36, 1115–1119. [Google Scholar] [PubMed]
- Duerinckx, S.; Meuwissen, M.; Perazzolo, C.; Desmyter, L.; Pirson, I.; Abramowicz, M. Phenotypes in siblings with homozygous mutations of TRAPPC9 and/or MCPH1 support a bifunctional model of MCPH1. Mol. Genet. Genom. Med. 2018, 6, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Sacher, M.; Kim, Y.G.; Lavie, A.; Oh, B.H.; Segev, N. The TRAPP complex: Insights into its architecture and function. Traffic 2008, 9, 2032–2042. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, S.E.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Singgih, E.L.; van Rens, A.; Demontis, D.; Børglum, A.D.; Mota, N.R.; Castells-Nobau, A.; Kiemeney, L.A.; Brunner, H.G.; Arias-Vasquez, A.; et al. Contribution of Intellectual Disability-Related Genes to ADHD Risk and to Locomotor Activity in Drosophila. Am. J. Psychiatry 2020, 177, 526–536. [Google Scholar] [CrossRef]
- Wanke, K.A.; Devanna, P.; Vernes, S.C. Understanding Neurodevelopmental Disorders: The Promise of Regulatory Variation in the 3′UTRome. Biol. Psychiatry 2018, 83, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Takata, A. Estimating contribution of rare non-coding variants to neuropsychiatric disorders. Psychiatry Clin. Neurosci. 2019, 73, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Barr, C.L.; Misener, V.L. Decoding the non-coding genome: Elucidating genetic risk outside the coding genome. Genes Brain Behav. 2016, 15, 187–204. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Mora, M.I.; Corominas, J.; Gilissen, C.; Sanchez, A.; Madrigal, I.; Rodriguez-Revenga, L. Novel Compound Heterozygous Mutation in TRAPPC9 Gene: The Relevance of Whole Genome Sequencing. Genes 2021, 12, 557. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040557
Alvarez-Mora MI, Corominas J, Gilissen C, Sanchez A, Madrigal I, Rodriguez-Revenga L. Novel Compound Heterozygous Mutation in TRAPPC9 Gene: The Relevance of Whole Genome Sequencing. Genes. 2021; 12(4):557. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040557
Chicago/Turabian StyleAlvarez-Mora, Maria Isabel, Jordi Corominas, Christian Gilissen, Aurora Sanchez, Irene Madrigal, and Laia Rodriguez-Revenga. 2021. "Novel Compound Heterozygous Mutation in TRAPPC9 Gene: The Relevance of Whole Genome Sequencing" Genes 12, no. 4: 557. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040557