
Growth Restriction and Genomic Imprinting-Overlapping Phenotypes Support the Concept of an Imprinting Network

 ,
,
Abstract
:1. Introduction
2. Imprinting Disorders
3. Factors Causing Aberrant Imprinting
3.1. Maternal Determinants with an Impact on Imprinting
3.2. Foetal Determinants with an Impact on Imprinting
3.3. The Imprinted Genes Network/IGN
4. Growth Restriction and Imprinting Disorders
5. Imprinting Disorders Associated with Growth Restriction
6. Transient Neonatal Diabetes Mellitus 1 (TNDM 1; 6q24)
6.1. Molecular Characteristics
6.2. Clinical Characteristics, Diagnosis and Therapy
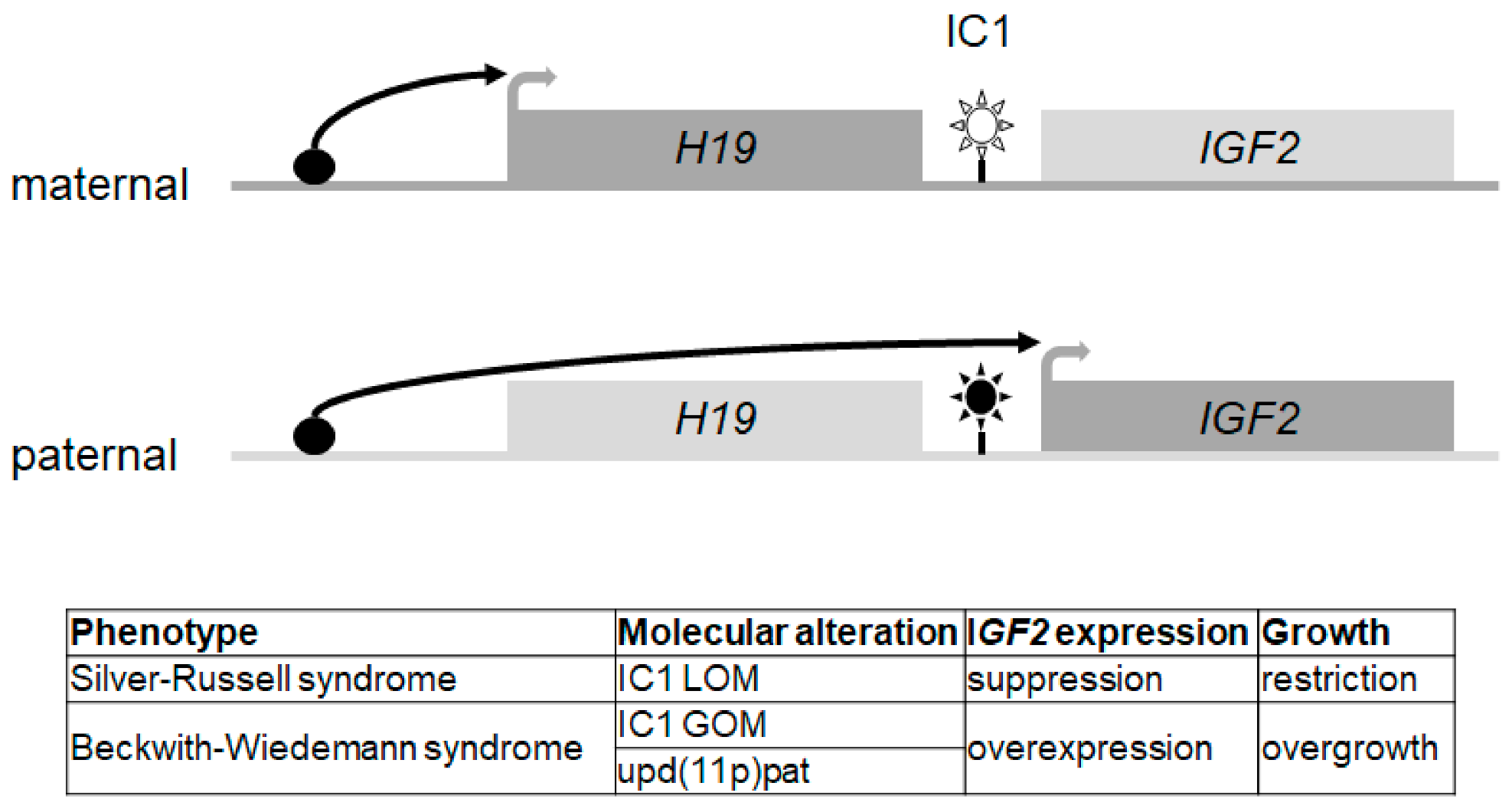
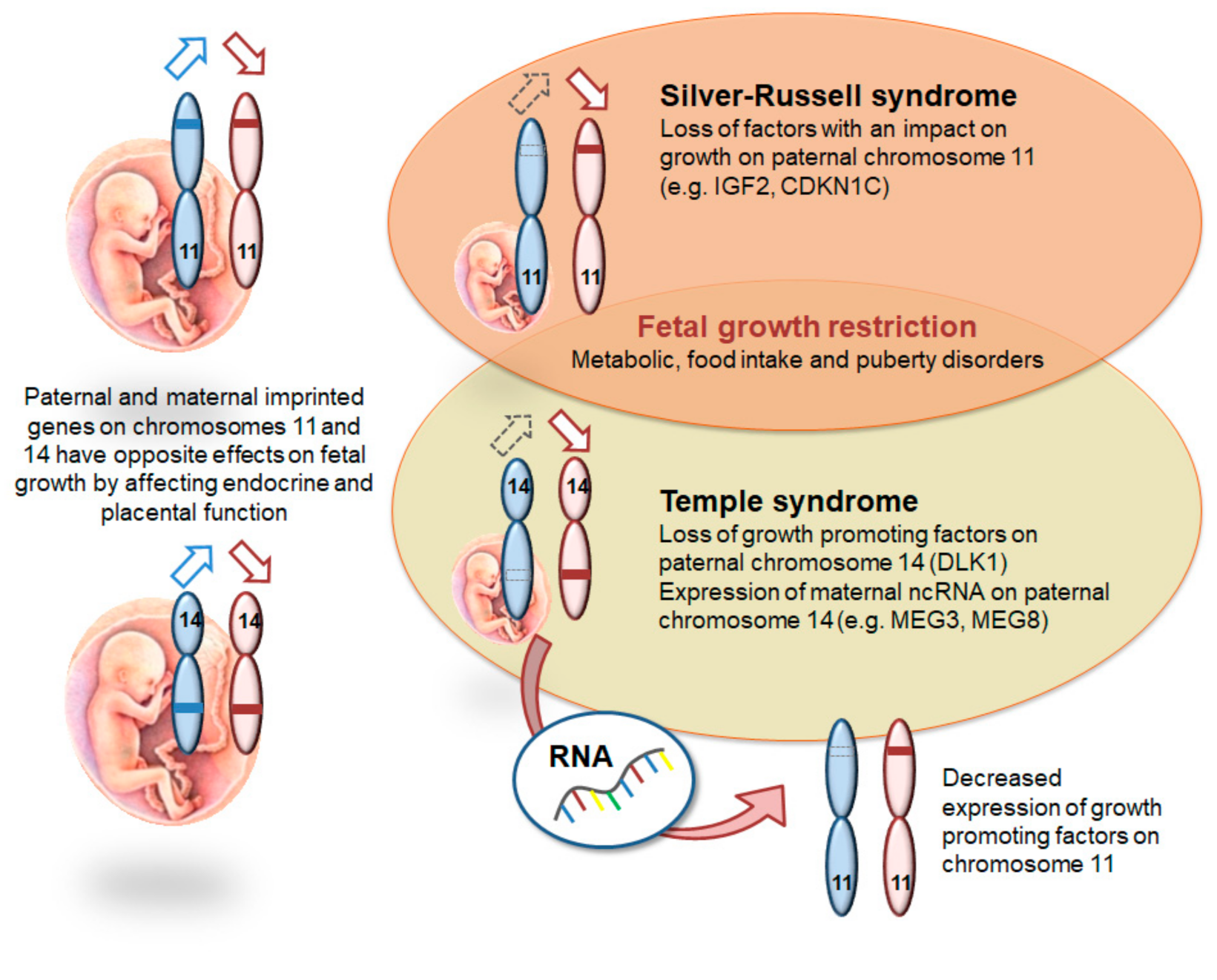
7. Silver-Russell Syndrome (SRS; 7p12, 7q32, 11p15.5)
7.1. Molecular Characteristics
7.2. Clinical Diagnosis and Therapy
8. Temple Syndrome (TS14; 14q32), (Central Precocious Puberty, CPPB)
8.1. Molecular Characteristics
8.2. Clinical Diagnosis and Therapy
9. Prader-Willi Syndrome (PWS, 15q11q13)
9.1. Molecular Characteristics
9.2. Clinical Diagnosis and Therapy
10. Pseudoparahypoparathyreoidism/Inactivating PTH/PTHrP Signalling Disorders (20q13.32)
10.1. Molecular Characteristics
10.2. Clinical Diagnosis and Therapy
11. Mulchandani–Bhoj–Conlin Syndrome (MBCS; 20q11q13)
11.1. Molecular Characteristics
11.2. Clinical Diagnosis and Therapy
12. Upd(6)mat and Upd(16)mat: Further Imprinting Disorders?
13. Translation and Transition
14. Conclusions and Outlook
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5mC | 5-methylcytosine |
| ART | Assisted reproduction technology |
| BWS | Beckwith–Wiedemann syndrome |
| CNVs | Copy number variants |
| DMR | Differentially methylated region (for nomenclature see: [7]) |
| GOM | Gain of methylation |
| IGN | Imprinted gene network |
| KOS14 | Kagami–Ogata syndrome |
| LOM | Loss of methylation |
| MILID | Multilocus imprinting disturbances |
| rGH | recombinant growth hormone |
| SCMC | Subcortical maternal complex |
| SGA | Small for gestational age |
| SNV | Single nucleotide variants |
| SRS | Silver-Russell syndrome |
| TS14 | Temple syndrome |
| UPD | Uniparental disomy |
References
- Gabory, A.; Ripoche, M.A.; Le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forne, T.; Jammes, H.; Ainscough, J.F.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillot-Durand, L.; Brioude, F.; Beneteau, C.; Le Breton, F.; Massardier, J.; Michon, L.; Devouassoux-Shisheboran, M.; Allias, F. Placental Pathology in Beckwith-Wiedemann Syndrome According to Genotype/Epigenotype Subgroups. Fetal Pediatr. Pathol. 2018, 37, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Plasschaert, R.N.; Bartolomei, M.S. Genomic imprinting in development, growth, behavior and stem cells. Development 2014, 141, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Soellner, L.; Begemann, M.; Mackay, D.J.; Gronskov, K.; Tumer, Z.; Maher, E.R.; Temple, I.K.; Monk, D.; Riccio, A.; Linglart, A.; et al. Recent Advances in Imprinting Disorders. Clin. Genet. 2017, 91, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Monk, D.; Morales, J.; den Dunnen, J.T.; Russo, S.; Court, F.; Prawitt, D.; Eggermann, T.; Beygo, J.; Buiting, K.; Tumer, Z.; et al. Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics 2018, 13, 117–121. [Google Scholar] [CrossRef]
- Docherty, L.E.; Kabwama, S.; Lehmann, A.; Hawke, E.; Harrison, L.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Shield, J.P.H.; Ennis, S.; et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype-phenotype correlation in an international cohort of patients. Diabetologia 2013, 56, 758–762. [Google Scholar] [CrossRef]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Kagami, M.; Nagasaki, K.; Kosaki, R.; Horikawa, R.; Naiki, Y.; Saitoh, S.; Tajima, T.; Yorifuji, T.; Numakura, C.; Mizuno, S.; et al. Temple syndrome: Comprehensive molecular and clinical findings in 32 Japanese patients. Genet. Med. 2017, 19, 1356–1366. [Google Scholar] [CrossRef] [Green Version]
- Ioannides, Y.; Lokulo-Sodipe, K.; Mackay, D.J.; Davies, J.H.; Temple, I.K. Temple syndrome: Improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: An analysis of 51 published cases. J. Med. Genet. 2014, 51, 495–501. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. Speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with, P.W.S. Recommendations for the diagnosis and management of Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef]
- Mantovani, G.; Bastepe, M.; Monk, D.; de Sanctis, L.; Thiele, S.; Usardi, A.; Ahmed, S.F.; Bufo, R.; Choplin, T.; De Filippo, G.; et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: First international Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 476–500. [Google Scholar] [CrossRef]
- Mulchandani, S.; Bhoj, E.J.; Luo, M.; Powell-Hamilton, N.; Jenny, K.; Gripp, K.W.; Elbracht, M.; Eggermann, T.; Turner, C.L.; Temple, I.K.; et al. Maternal uniparental disomy of chromosome 20: A novel imprinting disorder of growth failure. Genet. Med. 2016, 18, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, S.; Nakamura, A.; Inoue, T.; Matsubara, K.; Horikawa, R.; Wakui, K.; Takano, K.; Fukushima, Y.; Tatematsu, T.; Mizuno, S.; et al. Maternal Uniparental Disomy for Chromosome 20: Physical and Endocrinological Characteristics of Five Patients. J. Clin. Endocrinol. Metab. 2018, 103, 2083–2088. [Google Scholar] [CrossRef] [Green Version]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef]
- Kitsiou-Tzeli, S.; Tzetis, M. Maternal epigenetics and fetal and neonatal growth. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 43–46. [Google Scholar] [CrossRef]
- Taniguchi, K.; Kawai, T.; Hata, K. Placental Development and Nutritional Environment. Adv. Exp. Med. Biol. 2018, 1012, 63–73. [Google Scholar] [CrossRef]
- He, Q.L.; Wei, X.Y.; Han, X.Y.; Zhou, Q.; Wang, H.Q.; Ding, N.Z.; Meng, X.Q.; Schatten, H.; Sun, Q.Y.; Liu, S.Z. Effects of 2,3’,4,4’5-pentachlorobiphenyl exposure during pregnancy on epigenetic imprinting and maturation of offspring’s oocytes in mice. Arch. Toxicol. 2019, 93, 2575–2592. [Google Scholar] [CrossRef]
- Rhon-Calderon, E.A.; Vrooman, L.A.; Riesche, L.; Bartolomei, M.S. The effects of Assisted Reproductive Technologies on genomic imprinting in the placenta. Placenta 2019, 84, 37–43. [Google Scholar] [CrossRef]
- Mackay, D.J.; Eggermann, T.; Buiting, K.; Garin, I.; Netchine, I.; Linglart, A.; de Nanclares, G.P. Multilocus methylation defects in imprinting disorders. Biomol. Concepts 2015, 6, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Elbracht, M.; Mackay, D.; Begemann, M.; Kagan, K.O.; Eggermann, T. Disturbed genomic imprinting and its relevance for human reproduction: Causes and clinical consequences. Hum. Reprod. Update 2020, 26, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Valente, F.M.; Sparago, A.; Freschi, A.; Hill-Harfe, K.; Maas, S.M.; Frints, S.G.M.; Alders, M.; Pignata, L.; Franzese, M.; Angelini, C.; et al. Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith-Wiedemann locus. Genet. Med. 2019, 21, 1808–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beygo, J.; Citro, V.; Sparago, A.; De Crescenzo, A.; Cerrato, F.; Heitmann, M.; Rademacher, K.; Guala, A.; Enklaar, T.; Anichini, C.; et al. The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum. Mol. Genet. 2013, 22, 544–557. [Google Scholar] [CrossRef]
- Abi Habib, W.; Azzi, S.; Brioude, F.; Steunou, V.; Thibaud, N.; Das Neves, C.; Le Jule, M.; Chantot-Bastaraud, S.; Keren, B.; Lyonnet, S.; et al. Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 2014, 23, 5763–5773. [Google Scholar] [CrossRef] [Green Version]
- Boonen, S.E.; Porksen, S.; Mackay, D.J.; Oestergaard, E.; Olsen, B.; Brondum-Nielsen, K.; Temple, I.K.; Hahnemann, J.M. Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings. Eur. J. Hum. Genet. 2008, 16, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; Le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [CrossRef]
- Patten, M.M.; Cowley, M.; Oakey, R.J.; Feil, R. Regulatory links between imprinted genes: Evolutionary predictions and consequences. Proc. Biol. Sci. 2016, 283. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef]
- Abi Habib, W.; Brioude, F.; Azzi, S.; Rossignol, S.; Linglart, A.; Sobrier, M.L.; Giabicani, E.; Steunou, V.; Harbison, M.D.; Le Bouc, Y.; et al. Transcriptional profiling at the DLK1/MEG3 domain explains clinical overlap between imprinting disorders. Sci. Adv. 2019, 5, eaau9425. [Google Scholar] [CrossRef] [Green Version]
- Millership, S.J.; Van de Pette, M.; Withers, D.J. Genomic imprinting and its effects on postnatal growth and adult metabolism. Cell Mol. Life Sci. 2019, 76, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Monk, D.; Arnaud, P.; Frost, J.; Hills, F.A.; Stanier, P.; Feil, R.; Moore, G.E. Reciprocal imprinting of human GRB10 in placental trophoblast and brain: Evolutionary conservation of reversed allelic expression. Hum. Mol. Genet. 2009, 18, 3066–3074. [Google Scholar] [CrossRef]
- Masunaga, Y.; Inoue, T.; Yamoto, K.; Fujisawa, Y.; Sato, Y.; Kawashima-Sonoyama, Y.; Morisada, N.; Iijima, K.; Ohata, Y.; Namba, N.; et al. IGF2 Mutations. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef]
- Eggermann, T.; Binder, G.; Brioude, F.; Maher, E.R.; Lapunzina, P.; Cubellis, M.V.; Bergada, I.; Prawitt, D.; Begemann, M. CDKN1C mutations: Two sides of the same coin. Trends Mol. Med. 2014, 20, 614–622. [Google Scholar] [CrossRef]
- Abi Habib, W.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; Le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Temple, I.K.; Mackay, D.J.G. Diabetes Mellitus, 6q24-Related Transient Neonatal; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews((R)): Seattle, WA, USA, 1993. [Google Scholar]
- Mackay, D.J.; Callaway, J.L.; Marks, S.M.; White, H.E.; Acerini, C.L.; Boonen, S.E.; Dayanikli, P.; Firth, H.V.; Goodship, J.A.; Haemers, A.P.; et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 2008, 40, 949–951. [Google Scholar] [CrossRef]
- Beltrand, J.; Busiah, K.; Vaivre-Douret, L.; Fauret, A.L.; Berdugo, M.; Cave, H.; Polak, M. Neonatal Diabetes Mellitus. Front. Pediatr. 2020, 8, 540718. [Google Scholar] [CrossRef]
- Azzi, S.; Salem, J.; Thibaud, N.; Chantot-Bastaraud, S.; Lieber, E.; Netchine, I.; Harbison, M.D. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J. Med. Genet. 2015, 52, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Smeets, C.C.; Zandwijken, G.R.; Renes, J.S.; Hokken-Koelega, A.C. Long-Term Results of GH Treatment in Silver-Russell Syndrome (SRS): Do They Benefit the Same as Non-SRS Short-SGA? J. Clin. Endocrinol. Metab. 2016, 101, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Smeets, C.C.; Renes, J.S.; van der Steen, M.; Hokken-Koelega, A.C. Metabolic Health and Long-Term Safety of Growth Hormone Treatment in Silver-Russell Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 983–991. [Google Scholar] [CrossRef]
- Geoffron, S.; Abi Habib, W.; Chantot-Bastaraud, S.; Dubern, B.; Steunou, V.; Azzi, S.; Afenjar, A.; Busa, T.; Pinheiro Canton, A.; Chalouhi, C.; et al. Chromosome 14q32.2 Imprinted Region Disruption as an Alternative Molecular Diagnosis of Silver-Russell Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 2436–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brightman, D.S.; Lokulo-Sodipe, O.; Searle, B.A.; Mackay, D.J.G.; Davies, J.H.; Temple, I.K.; Dauber, A. Growth Hormone Improves Short-Term Growth in Patients with Temple Syndrome. Horm. Res. Paediatr. 2018, 90, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Severi, G.; Bernardini, L.; Briuglia, S.; Bigoni, S.; Buldrini, B.; Magini, P.; Dentici, M.L.; Cordelli, D.M.; Arrigo, T.; Franzoni, E.; et al. New patients with Temple syndrome caused by 14q32 deletion: Genotype-phenotype correlations and risk of thyroid cancer. Am. J. Med. Genet. A 2016, 170A, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R.D.; Horsthemke, B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995, 9, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Cassidy, S.B.; Driscoll, D.J.; Gillessen-Kaesbach, G.; Kanber, D.; Tauber, M.; Schwinger, E.; Horsthemke, B. Clinical utility gene card for: Prader-Willi Syndrome. Eur. J. Hum. Genet. 2014, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buiting, K.; Clayton-Smith, J.; Driscoll, D.J.; Gillessen-Kaesbach, G.; Kanber, D.; Schwinger, E.; Williams, C.; Horsthemke, B. Clinical utility gene card for: Angelman Syndrome. Eur. J. Hum. Genet. 2015, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buiting, K.; Gross, S.; Lich, C.; Gillessen-Kaesbach, G.; el-Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Swaab, D.F. Prader-Willi syndrome and the hypothalamus. Acta Paediatr. Suppl. 1997, 423, 50–54. [Google Scholar] [CrossRef] [Green Version]
- de Lind van Wijngaarden, R.F.; de Klerk, L.W.; Festen, D.A.; Hokken-Koelega, A.C. Scoliosis in Prader-Willi syndrome: Prevalence, effects of age, gender, body mass index, lean body mass and genotype. Arch. Dis. Child. 2008, 93, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Tauber, M.; Hoybye, C. Endocrine disorders in Prader-Willi syndrome: A model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol. 2021. [Google Scholar] [CrossRef]
- Bakker, N.E.; Kuppens, R.J.; Siemensma, E.P.; Tummers-de Lind van Wijngaarden, R.F.; Festen, D.A.; Bindels-de Heus, G.C.; Bocca, G.; Haring, D.A.; Hoorweg-Nijman, J.J.; Houdijk, E.C.; et al. Eight years of growth hormone treatment in children with Prader-Willi syndrome: Maintaining the positive effects. J. Clin. Endocrinol. Metab. 2013, 98, 4013–4022. [Google Scholar] [CrossRef] [Green Version]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, G.; Bastepe, M.; Monk, D.; de Sanctis, L.; Thiele, S.; Ahmed, S.F.; Bufo, R.; Choplin, T.; De Filippo, G.; Devernois, G.; et al. Recommendations for Diagnosis and Treatment of Pseudohypoparathyroidism and Related Disorders: An Updated Practical Tool for Physicians and Patients. Horm. Res. Paediatr. 2020, 93, 182–196. [Google Scholar] [CrossRef]
- Hjortshoj, T.D.; Sorensen, A.R.; Yusibova, M.; Hansen, B.M.; Duno, M.; Balslev-Harder, M.; Gronskov, K.; van Hagen, J.M.; Polstra, A.M.; Eggermann, T.; et al. upd(20)mat is a rare cause of the Silver-Russell-syndrome-like phenotype: Two unrelated cases and screening of large cohorts. Clin. Genet. 2020, 97, 902–907. [Google Scholar] [CrossRef]
- Scheuvens, R.; Begemann, M.; Soellner, L.; Meschede, D.; Raabe-Meyer, G.; Elbracht, M.; Schubert, R.; Eggermann, T. Maternal uniparental disomy of chromosome 16 [upd(16)mat]: Clinical features are rather caused by (hidden) trisomy 16 mosaicism than by upd(16)mat itself. Clin. Genet. 2017, 92, 45–51. [Google Scholar] [CrossRef]
- Inoue, T.; Yagasaki, H.; Nishioka, J.; Nakamura, A.; Matsubara, K.; Narumi, S.; Nakabayashi, K.; Yamazawa, K.; Fuke, T.; Oka, A.; et al. Molecular and clinical analyses of two patients with UPD(16)mat detected by screening 94 patients with Silver-Russell syndrome phenotype of unknown aetiology. J. Med. Genet. 2019, 56, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Eggermann, T.; Oehl-Jaschkowitz, B.; Dicks, S.; Thomas, W.; Kanber, D.; Albrecht, B.; Begemann, M.; Kurth, I.; Beygo, J.; Buiting, K. The maternal uniparental disomy of chromosome 6 (upd(6)mat) “phenotype”: Result of placental trisomy 6 mosaicism? Mol. Genet. Genomic Med. 2017, 5, 668–677. [Google Scholar] [CrossRef] [Green Version]
- Salem, J.B.; Netchine, I.; Harbison, M.D. The Importance of Collaboration in Advancing Understanding of Rare Disorders: US/EU Joint Initiative on Silver-Russell Syndrome. Pediatr. Endocrinol. Rev. 2017, 15, 98–101. [Google Scholar] [CrossRef]
- Meyer, R.; Begemann, M.; Hubner, C.T.; Dey, D.; Kuechler, A.; Elgizouli, M.; Schara, U.; Ambrozaityte, L.; Burnyte, B.; Schroder, C.; et al. One test for all: Whole exome sequencing significantly improves the diagnostic yield in growth retarded patients referred for molecular testing for Silver-Russell syndrome. Orphanet. J. Rare Dis. 2021, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 325. [Google Scholar] [CrossRef] [PubMed]
- Paepegaey, A.C.; Coupaye, M.; Jaziri, A.; Menesguen, F.; Dubern, B.; Polak, M.; Oppert, J.M.; Tauber, M.; Pinto, G.; Poitou, C. Impact of transitional care on endocrine and anthropometric parameters in Prader-Willi syndrome. Endocr. Connect. 2018, 7, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Lokulo-Sodipe, O.; Ballard, L.; Child, J.; Inskip, H.M.; Byrne, C.D.; Ishida, M.; Moore, G.E.; Wakeling, E.L.; Fenwick, A.; Mackay, D.J.G.; et al. Phenotype of genetically confirmed Silver-Russell syndrome beyond childhood. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Liao, J.; Zeng, T.B.; Pierce, N.; Tran, D.A.; Singh, P.; Mann, J.R.; Szabo, P.E. Prenatal correction of IGF2 to rescue the growth phenotypes in mouse models of Beckwith-Wiedemann and Silver-Russell syndromes. Cell Rep. 2021, 34, 108729. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Imprinting Disorder OMIM | Prevalence | Chromosome | Molecular Defect (Frequency) | MLID | Main Clinical Features | References |
|---|---|---|---|---|---|---|
| Transient neonatal diabetes mellitus (TNDM) 601410 | 1/300.000 | 6q24 |
| 30% | IUGR, transient diabetes mellitus, hyperglycaemia without ketoacidosis, macroglossia, abdominal wall defects | [8] |
| Silver-Russell syndrome (SRS) 180860 | 1/75.000–1/100.000 | Chr 7 Chr 11p15 |
| 1 case 7–10% | IUGR, PNGR, relative macrocephaly at birth, body asymmetry (11p15), prominent forehead, feeding difficulties in early childhood | [9] |
| Temple syndrome (TS14) 616222 | unknown | Chr 14q32 |
| ? | IUGR, PNGR, neonatal hypotonia, feeding difficulties in infancy, truncal obesity, scoliosis, precocious puberty, small feet and hands | [10,11] |
| Prader-Willi syndrome (PWS) 176270 | 1/25.000 –1/15.000 | Chr 15q11.2 |
| 1 case? | Neonatal hypotonia, severe feeding difficulties in infancy (poor suck), hypogenitalism, PNGR, psychomotor developmental delay, intellectual disability, behavioural problems (tantrums), hyperphagia, (extreme) obesity, hypogonadism, hypopigmentation, scoliosis, abnormal pubertal progression small hands and feet | [12] |
| Pseudohypo-parathyroidism 1B (PHP1B) 603233 | Unknown | Chr 20q13 |
| 12.5% | macrosomia, PTH resistance, TSH resistance, Albright hereditary osteodystrophy, early onset obesity subcutaneous ossifications | [13] |
| Mulchandani-Bhoj-Conlin syndrome (MBCS) 617352 | Unknown | Chr 20 |
| IUGR, PNGR, microcephaly, feeding difficulties, psychomotor developmental delay in some children | [14,15] |
| Imprinting Disorder | OMIM | Chromosome | Imprinted Genes in the Region a | IUGR | PNGR | Foetal Macrosomia | Postnatal Overgrowth | (Relative) Macrocephaly | Asymmetry | Abdominal Wall Defects | Macroglossia | Metabolic Disturbance | Feeding Difficulties in Infancy | (Truncal) Obesity | Scoliosis | Early or Precocious Puberty | (Neonatal) Hypotonia | Ccognitive Impairment | Embryonal Tumor | Placental Pathology | Polyhydramnios |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transient neonatal diabetes mellitus (TNDM) | 601410 | 6q24 | PLAGL1 | yes | yes | Yes | hyperglycemia without ketoacidosis | ||||||||||||||
| Silver-Russell syndrome b (SRS) | 180860 | 11p15.5 | IGF2, CDKN1C | yes | yes | yes | yes | hypoglycaemia/ insulin resistance in young adults | yes | yes c | yes | yes | |||||||||
| 618905 | 7p13q32 | GRB10, MEST | yes | yes | yes | yes | hypoglycaemia | yes | yes | yes | yes | yes d | |||||||||
| Birk-Barel syndrome | 612292 | 8q24.3 | KCNK9 | yes | yes | ||||||||||||||||
| Beckwith–Wiedemann syndrome (BWS) | 130650 | 11p15.5 | CDKN1C, IGF2, H19 | yes | yes | yes | yes | Yes | hyperinsulinism | yes | yes | yes | |||||||||
| Temple syndrome (TS14) | 616222 | 14q32 | DLK1 | yes | yes | yes | yes | insulin resistance | yes | yes | yes | yes | yes | yes | |||||||
| Kagami–Ogata syndrome (KOS14) | 608149 | 14q32 | DLK1 | yes | yes | yes | yes | yes | yes | yes | yes | yes | |||||||||
| (familial) central precocious puberty (CBBP) | (DLK1: 176290) | 14q32 | DLK1 | yes | |||||||||||||||||
| Prader-Willi syndrome (PWS) | 176270 | 15q11.2 | SNRPN | yes | yes | no | facial asymmetry in neoates | hypoinsulinemia and high insulin sensitivity | yes | yes | yes | yes | yes | ||||||||
| Angelman syndrome (AS) | 105830 | 15q11.2 | SNRPN, UBE3A | yes | yes | ||||||||||||||||
| Central precocious puberty 2 (CPPB2) | 615356 | 15q11.2 | MKRN3 | yes | |||||||||||||||||
| Schaaf-Yang syndrome (SYS) | 615547 | 15q11.2 | MAGEL2 | yes | hyperinsulinism | yes | yes | yes | |||||||||||||
| Pseudohypoparathyroidism 1B (PHP1b) | 603233 | 20q13 | GNAS complex | yes | yes | yes | yes | yes | yes | Yes | |||||||||||
| Mulchandani–Bhoj–Conlin syndrome (MBCS) | 617352 | 20 | yes | yes | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eggermann, T.; Davies, J.H.; Tauber, M.; van den Akker, E.; Hokken-Koelega, A.; Johansson, G.; Netchine, I. Growth Restriction and Genomic Imprinting-Overlapping Phenotypes Support the Concept of an Imprinting Network. Genes 2021, 12, 585. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040585
Eggermann T, Davies JH, Tauber M, van den Akker E, Hokken-Koelega A, Johansson G, Netchine I. Growth Restriction and Genomic Imprinting-Overlapping Phenotypes Support the Concept of an Imprinting Network. Genes. 2021; 12(4):585. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040585
Chicago/Turabian StyleEggermann, Thomas, Justin H. Davies, Maithé Tauber, Erica van den Akker, Anita Hokken-Koelega, Gudmundur Johansson, and Irène Netchine. 2021. "Growth Restriction and Genomic Imprinting-Overlapping Phenotypes Support the Concept of an Imprinting Network" Genes 12, no. 4: 585. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040585