
Maternal Uniparental Disomy of Chromosome 20 (UPD(20)mat) as Differential Diagnosis of Silver Russell Syndrome: Identification of Three New Cases

 , , , , , and
, , , , , and
Abstract
:1. Introduction
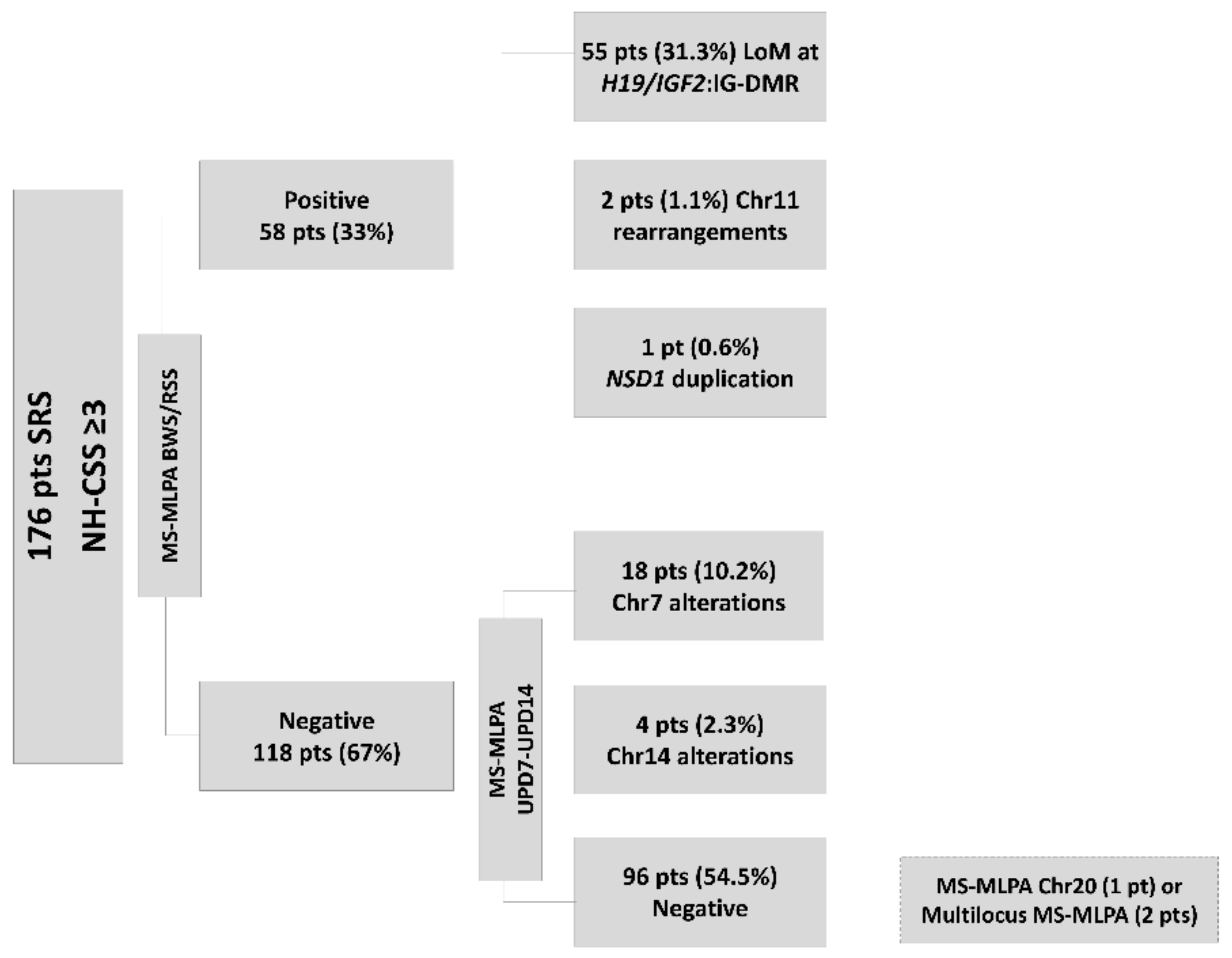
2. Materials and Methods
2.1. Population Study
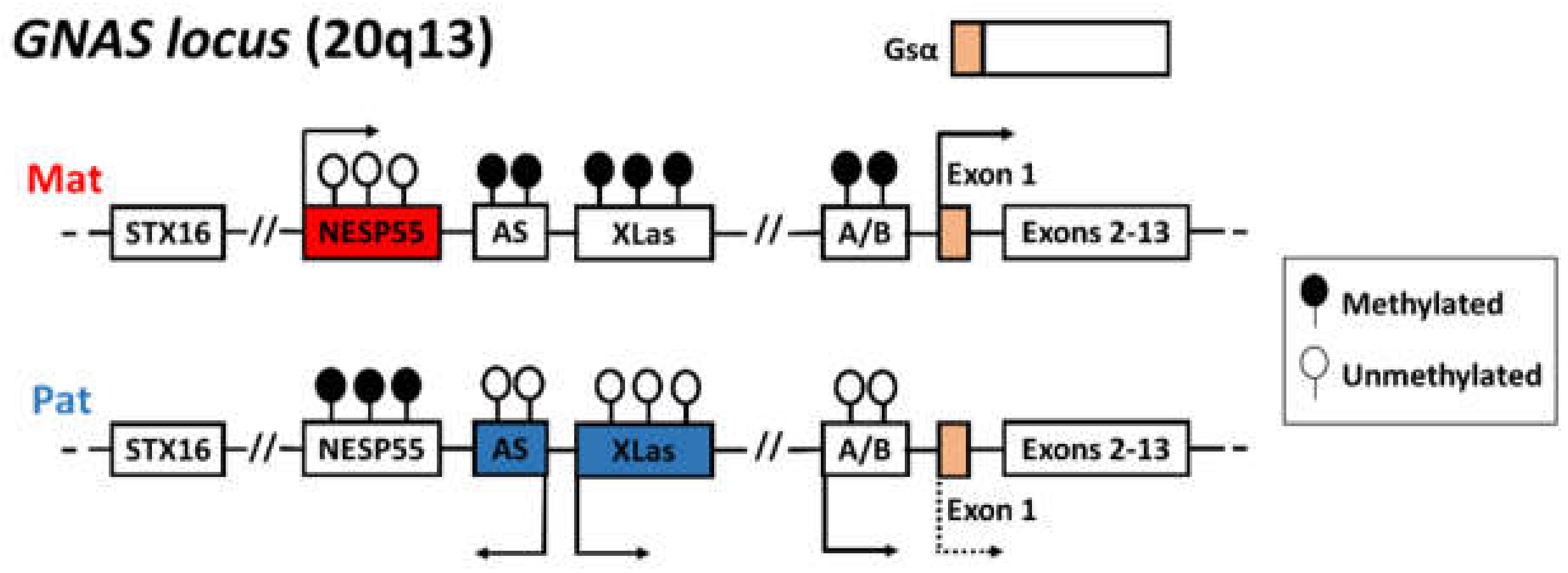
2.2. Molecular Analysis and Study Design
3. Results
3.1. UPD(20)mat Patients
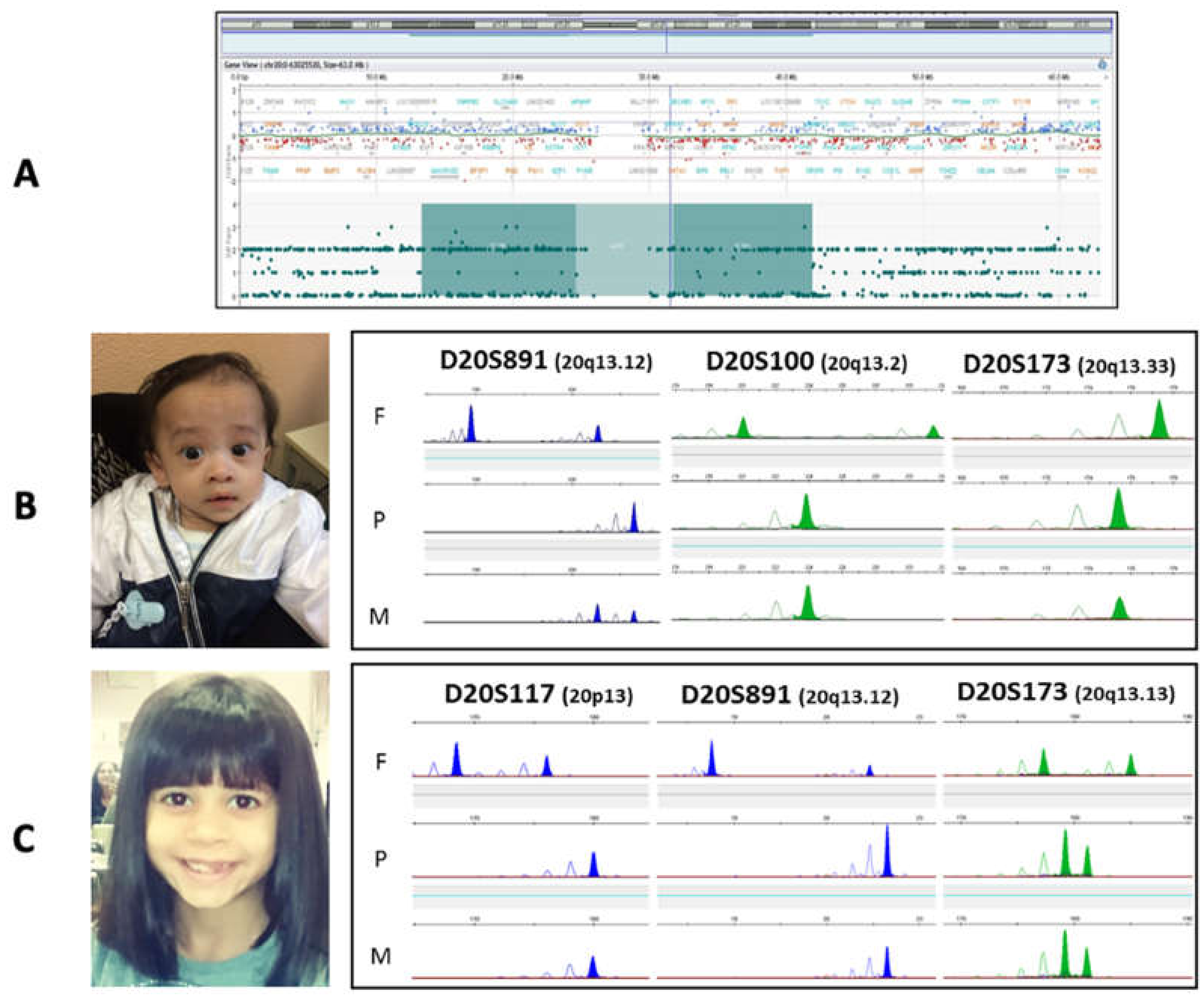
3.1.1. Patient 1
3.1.2. Patient 2
3.1.3. Patient 3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, F.; Sparago, A.; Ariani, F.; Brugnoletti, F.; Calzari, L.; Coppede, F.; De Luca, A.; Gervasini, C.; Giardina, E.; Gurrieri, F.; et al. DNA Methylation in the Diagnosis of Monogenic Diseases. Genes 2020, 11, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuke, T.; Nakamura, A.; Inoue, T.; Kawashima, S.; Hara, K.I.; Matsubara, K.; Sano, S.; Yamazawa, K.; Fukami, M.; Ogata, T.; et al. Role of imprinting disorders in short children born SGA and Silver-Russell syndrome spectrum. J. Clin. Endocrinol. Metab. 2020, 106, 802–813. [Google Scholar] [CrossRef]
- Eggermann, T.; Mackay, D.; Tümer, Z. Uniparental Disomy and Imprinting Disorders. OBM Genetics 2018, 2, 1. [Google Scholar] [CrossRef]
- Eggermann, T.; Begemann, M.; Kurth, I.; Elbracht, M. Contribution of GRB10 to the prenatal phenotype in Silver-Russell syndrome? Lessons from 7p12 copy number variations. Eur. J. Med. Genet. 2019, 62, 103671. [Google Scholar] [CrossRef]
- Carrera, I.A.; de Zaldivar, M.S.; Martin, R.; Begemann, M.; Soellner, L.; Eggermann, T. Microdeletions of the 7q32.2 imprinted region are associated with Silver-Russell syndrome features. Am. J. Med Genet. Part A 2016, 170, 743–749. [Google Scholar] [CrossRef]
- Crippa, M.; Bonati, M.T.; Calzari, L.; Picinelli, C.; Gervasini, C.; Sironi, A.; Bestetti, I.; Guzzetti, S.; Bellone, S.; Selicorni, A.; et al. Molecular Etiology Disclosed by Array CGH in Patients with Silver-Russell Syndrome or Similar Phenotypes. Front. Genet. 2019, 10, 955. [Google Scholar] [CrossRef] [Green Version]
- Abi Habib, W.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; Le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Chudoba, I.; Franke, Y.; Senger, G.; Sauerbrei, G.; Demuth, S.; Beensen, V.; Neumann, A.; Hansmann, I.; Claussen, U. Maternal UPD 20 in a hyperactive child with severe growth retardation. Eur. J. Hum. Genet. 1999, 7, 533–540. [Google Scholar] [CrossRef]
- Eggermann, T.; Mergenthaler, S.; Eggermann, K.; Albers, A.; Linnemann, K.; Fusch, C.; Ranke, M.B.; Wollmann, H.A. Identification of interstitial maternal uniparental disomy (UPD) (14) and complete maternal UPD(20) in a cohort of growth retarded patients. J. Med. Genet. 2001, 38, 86–89. [Google Scholar] [CrossRef]
- Salafsky, I.S.; MacGregor, S.N.; Claussen, U.; von Eggeling, F. Maternal UPD 20 in an infant from a pregnancy with mosaic trisomy 20. Prenat. Diagn. 2001, 21, 860–863. [Google Scholar] [CrossRef]
- Velissariou, V.; Antoniadi, T.; Gyftodimou, J.; Bakou, K.; Grigoriadou, M.; Christopoulou, S.; Hatzipouliou, A.; Donoghue, J.; Karatzis, P.; Katsarou, E.; et al. Maternal uniparental isodisomy 20 in a foetus with trisomy 20 mosaicism: Clinical, cytogenetic and molecular analysis. Eur. J. Hum. Genet. 2002, 10, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Azzi, S.; Salem, J.; Thibaud, N.; Chantot-Bastaraud, S.; Lieber, E.; Netchine, I.; Harbison, M.D. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J. Med. Genet. 2015, 52, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Mulchandani, S.; Bhoj, E.J.; Luo, M.; Powell-Hamilton, N.; Jenny, K.; Gripp, K.W.; Elbracht, M.; Eggermann, T.; Turner, C.L.; Temple, I.K.; et al. Maternal uniparental disomy of chromosome 20: A novel imprinting disorder of growth failure. Genet. Med. 2016, 18, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, S.; Nakamura, A.; Inoue, T.; Matsubara, K.; Horikawa, R.; Wakui, K.; Takano, K.; Fukushima, Y.; Tatematsu, T.; Mizuno, S.; et al. Maternal Uniparental Disomy for Chromosome 20: Physical and Endocrinological Characteristics of Five Patients. J. Clin. Endocrinol. Metab. 2018, 103, 2083–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajan, S.A.; Powis, Z.; Helbig, K.L.; Nagakura, H.; Immken, L.; Tang, S.; Alcaraz, W.A. Diagnostic exome sequencing identifies GLI2 haploinsufficiency and chromosome 20 uniparental disomy in a patient with developmental anomalies. Clin. Case Rep. 2018, 6, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Hjortshoj, T.D.; Sorensen, A.R.; Yusibova, M.; Hansen, B.M.; Duno, M.; Balslev-Harder, M.; Gronskov, K.; van Hagen, J.M.; Polstra, A.M.; Eggermann, T.; et al. upd(20)mat is a rare cause of the Silver-Russell-syndrome-like phenotype: Two unrelated cases and screening of large cohorts. Clin. Genet. 2020, 97, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.; Grybek, V.; Perez de Nanclares, G.; Tran, L.C.; de Sanctis, L.; Elli, F.; Errea, J.; Francou, B.; Kamenicky, P.; Linglart, L.; et al. Genetic and Epigenetic Defects at the GNAS Locus Lead to Distinct Patterns of Skeletal Growth but Similar Early-Onset Obesity. J. Bone Miner. Res. 2018, 33, 1480–1488. [Google Scholar] [CrossRef]
- Bastepe, M.; Juppner, H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005, 63, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, G.; Bastepe, M.; Monk, D.; de Sanctis, L.; Thiele, S.; Usardi, A.; Ahmed, S.F.; Bufo, R.; Choplin, T.; De Filippo, G.; et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: First international Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 476–500. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Gavrilova, O.; Chen, H.; Lee, R.; Liu, J.; Pacak, K.; Parlow, A.F.; Quon, M.J.; Reitman, M.L.; Weinstein, L.S. Paternal versus maternal transmission of a stimulatory G-protein α subunit knockout produces opposite effects on energy metabolism. J. Clin. Investig. 2000, 105, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Plagge, A.; Gordon, E.; Dean, W.; Boiani, R.; Cinti, S.; Peters, J.; Kelsey, G. The imprinted signaling protein XL α s is required for postnatal adaptation to feeding. Nat. Genet. 2004, 36, 818–826. [Google Scholar] [CrossRef]
- Xie, T.; Plagge, A.; Gavrilova, O.; Pack, S.; Jou, W.; Lai, E.W.; Frontera, M.; Kelsey, G.; Weinstein, L.S. The alternative stimulatory G protein α-subunit XLalphas is a critical regulator of energy and glucose metabolism and sympathetic nerve activity in adult mice. J. Biol. Chem. 2006, 281, 18989–18999. [Google Scholar] [CrossRef] [Green Version]
- Long, D.N.; McGuire, S.; Levine, M.A.; Weinstein, L.S.; Germain-Lee, E.L. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Galpha(s) in the development of human obesity. J. Clin. Endocrinol. Metab. 2007, 92, 1073–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallerstein, R.; Yu, M.T.; Neu, R.L.; Benn, P.; Lee Bowen, C.; Crandall, B.; Disteche, C.; Donahue, R.; Harrison, B.; Hershey, D.; et al. Common trisomy mosaicism diagnosed in amniocytes involving chromosomes 13, 18, 20 and 21: Karyotype-phenotype correlations. Prenat. Diagn. 2000, 20, 103–122. [Google Scholar] [CrossRef]
- Powis, Z.; Erickson, R.P. Uniparental disomy and the phenotype of mosaic trisomy 20: A new case and review of the literature. J. Appl. Genet. 2009, 50, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Ensenauer, R.E.; Shaughnessy, W.J.; Jalal, S.M.; Dawson, D.B.; Courteau, L.K.; Ellison, J.W. Trisomy 20 mosaicism caused by a maternal meiosis II error is associated with normal intellect but multiple congenital anomalies. Am. J. Med. Genet. Part A 2005, 134, 202–206. [Google Scholar] [CrossRef]
- Ball, S.T.; Kelly, M.L.; Robson, J.E.; Turner, M.D.; Harrison, J.; Jones, L.; Napper, D.; Beechey, C.V.; Hough, T.; Plagge, A.; et al. Gene Dosage Effects at the Imprinted Gnas Cluster. PLoS ONE 2013, 8, e65639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constancia, M.; Kelsey, G.; Reik, W. Resourceful imprinting. Nature 2004, 432, 53–57. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Patient 1 | Patient 2 | Patient 3 | Previously Reported Cases (n = 21) |
|---|---|---|---|---|
| Sex | M | M | F | 11M/9F/1NA |
| Maternal age at birth (years) | 44 | 42 | 40 | 38 (28–43) |
| Gestational age (weeks + days) | 36 + 4 | 38 + 5 | 38 + 1 | 37 (32–40) |
| Oligohydramnios | NA | None | None | 6/18 |
| Birth length (cm | SDS) | 43 | −2.27 | 44 | −2.65 | 46 | −1.7 | – |
| Birth weight (kg | SDS) | 1.51 | −3.12 | 2.23 | −2.38 | 2.48 | −1.79 | – |
| Birth head circumference (cm | SDS) | 30 | −2.53 | 33.1 | −1.01 | 31.5 | −1.96 | – |
| Age at clinical SRS suspicion (months) | 15 | 5.5 | 15 | – |
| Evaluation length (cm | SDS) | 71.5 | −2.7 | 60 | −2.7 | 69.9 | –2.5 | – |
| Evaluation weight (kg | SDS) | 6.7 | –4.9 | 4.15 | −5 | 6.52 | −4.9 | – |
| UPD type | NA | UPhD | UPhD + UPiD | 5/21 UPiD; 4/21 UPhD; 10/21 mixed |
| Methods | MS-MLPA + SNP-array | MS-MLPA + STRs | MS-MLPA + STRs | Various * |
| NH-CSS 1 features: | ||||
| SGA (birth weight and/or length ≤ –2 SDS) | + | + | – | 15/21 |
| Relative macrocephaly at birth a | – | + | – | 4/10 |
| Postnatal growth failure b | + # | +/–$ | + # | 18/21 # |
| Protruding forehead c | + | + | + | 4/8 |
| Body asymmetry d | + | – | – | 2/8 |
| Feeding difficulties e and/or BMI ≤ –2 SDS (2ys) | + | + | + | 18/21 |
| Other clinical manifestations: | ||||
| Small and triangular face | + | + | + | 6/8 |
| Micrognathia | + | + | – | 1/1 |
| Hypotonia | + | – | + | 9/13 |
| Developmental delay | – | – | – | 6/16 |
| GH deficit | + | – | + | 2/21 |
| Facial dysmorphism | Blue sclera, ear anomalies | Epicanthus, helix hypoplasia, short philtrum, thin lips | Short palpebral fissures | 11/21 |
| Skeletal abnormalities | None | Fifth finger clinodactyly | None | 13/19 (5/6 clinodactyly) |
| Genital anomalies | Cryptorchidism | None | Vaginal synechiae | 3/3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tannorella, P.; Minervino, D.; Guzzetti, S.; Vimercati, A.; Calzari, L.; Patti, G.; Maghnie, M.; Allegri, A.E.M.; Milani, D.; Scuvera, G.; et al. Maternal Uniparental Disomy of Chromosome 20 (UPD(20)mat) as Differential Diagnosis of Silver Russell Syndrome: Identification of Three New Cases. Genes 2021, 12, 588. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040588
Tannorella P, Minervino D, Guzzetti S, Vimercati A, Calzari L, Patti G, Maghnie M, Allegri AEM, Milani D, Scuvera G, et al. Maternal Uniparental Disomy of Chromosome 20 (UPD(20)mat) as Differential Diagnosis of Silver Russell Syndrome: Identification of Three New Cases. Genes. 2021; 12(4):588. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040588
Chicago/Turabian StyleTannorella, Pierpaola, Daniele Minervino, Sara Guzzetti, Alessandro Vimercati, Luciano Calzari, Giuseppa Patti, Mohamad Maghnie, Anna Elsa Maria Allegri, Donatella Milani, Giulietta Scuvera, and et al. 2021. "Maternal Uniparental Disomy of Chromosome 20 (UPD(20)mat) as Differential Diagnosis of Silver Russell Syndrome: Identification of Three New Cases" Genes 12, no. 4: 588. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040588