
Influence of Age on Skeletal Muscle Hypertrophy and Atrophy Signaling: Established Paradigms and Unexpected Links


Department of Orthopaedic Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
*
Author to whom correspondence should be addressed.
Genes 2021, 12(5), 688; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050688
Submission received: 2 April 2021
/
Revised: 26 April 2021
/
Accepted: 27 April 2021
/
Published: 3 May 2021
(This article belongs to the Special Issue Genetic and Environmental Factors in Ageing and Age-Related Disease)
{kind=link}
{kind=link}
Abstract
:Skeletal muscle atrophy in an inevitable occurrence with advancing age, and a consequence of disease including cancer. Muscle atrophy in the elderly is managed by a regimen of resistance exercise and increased protein intake. Understanding the signaling that regulates muscle mass may identify potential therapeutic targets for the prevention and reversal of muscle atrophy in metabolic and neuromuscular diseases. This review covers the major anabolic and catabolic pathways that regulate skeletal muscle mass, with a focus on recent progress and potential new players.
1. Introduction
Biological aging is broadly defined as the time-dependent loss of functionality and robustness, and is associated with increased rates of cancer [1,2], cardiovascular and neurodegenerative disease [2,3,4], diabetes or dysglycemia [5], insulin and anabolic resistance [6,7,8]. Many of these disorders and diseases are often accompanied by, and contribute to, muscle weakness and sarcopenia, exacerbating the slow gradual loss of muscle mass and strength that typically begins in the fourth decade of life [9,10,11]. In healthy young individuals, lean muscle accounts for 38–54% and 28–39% of total body mass, in men and women, respectively. These ranges are quite broad and are dependent upon multiple factors including physical activity level, overall health, genetic makeup, and nutritional input. Skeletal muscle is comprised of a heterogenous mix of specialized myofibers that differ in their physiological, metabolic, and biochemical attributes. On the two ends of this spectrum are slow oxidative fibers, comprised of type I myosin heavy chain proteins (MHC), and the fast-glycolytic fibers, comprised of types IIa, IIx, and IIb (in rodents) MHC proteins. With advancing age, skeletal muscle loses its responsiveness to anabolic signals, which are secreted in ever diminishing levels [8,12,13,14]. Importantly, fast-glycolytic fibers atrophy at a greater rate than oxidative fibers with advancing age [15,16,17], contributing significantly to the loss of muscle power that begins its decline from approximately age 40 onwards [18]. It is this loss of muscle power that is related to the increased number of falls among the elderly [18,19].
The cellular and molecular mechanisms linking biological aging with the loss of muscle mass, strength, and functionality are complex. In its simplest form, the maintenance of muscle mass, and strength, is due to the balance of anabolic and catabolic processes. Stem cell exhaustion, cellular senescence, altered intercellular communication, losses of proteostasis and genome stability, telomere attrition, mitochondrial dysfunction, epigenetic alterations, and dysregulation of gene expression and alternative splicing are hallmarks of, and causal for, the age-associated loss of cellular and biological robustness [20,21,22,23]. In this review, we identify and discuss the cellular and molecular mechanisms responsible for modulating skeletal muscle mass and strength, and its influence on the non-myocyte contributions to skeletal muscle physiology that become dysregulated with age. Herein we give high priority to in vivo and clinical studies identifying the effects of age on the linkages between gene expression and signaling pathways with muscle function. Finally, we would like to sincerely apologize to our many outstanding colleagues, whose work may have been inadvertently overlooked or not thoroughly discussed due to space constraints.
2. PI3K/Akt/mTOR Signaling in Muscle
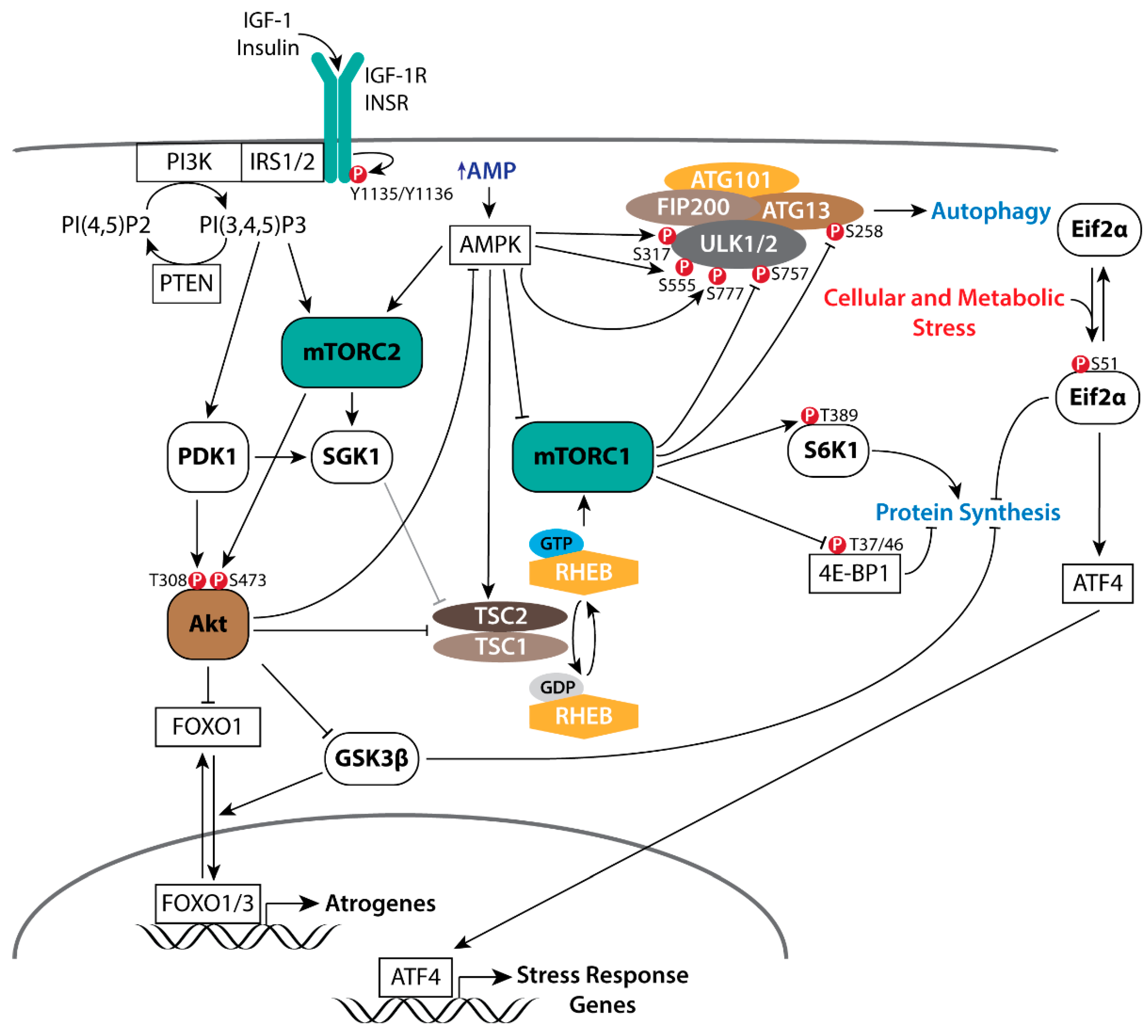
Insulin, and insulin-like growth factor 1 (IGF-1) signaling (IIS) is ubiquitous to multicellular animals, connecting nutrient availability to development, longevity, and growth in response to hypothalamic-pituitary growth signals [24]. Activation of the insulin/IGF-1 receptors leads to the phospho-activation of phosphoinositide 3-kinase (PI3K), and the subsequent activation of Akt (Figure 1), a key integrator of intra- and intercellular signals known to promote cell survival, glucose metabolism, and anabolic growth processes [25,26,27,28,29,30,31,32]. Importantly, IIS has evolutionarily conserved pleiotropic effects that are age/developmental stage and gender dependent. Ames, Snell, and GHRH (lit/lit) dwarf mice have reduced growth hormone (GH) signaling and reduced serum IGF-1 levels, but have up to a 70% extension of their lifespans [33,34,35,36,37,38]. Heterozygous deletion of the IGF-1 receptor gene (Igf1r) have a modest ~6–8% reduction in bodyweight by 7 weeks of age, but live up to 25% longer [39,40]. However, this lifespan extension was limited to females and modulated by genetic background [39,41,42]. Consistent with the influence of gender, 18-month-old female mice treated with a selective IGF-1R antagonist for 6 months, but not male mice, showed significant improvements in exercise tolerance, grip strength, and motor coordination [43]. In contrast, transgenic expression of GH in mice induces accelerated growth, hyperinsulinemia despite euglycemia, increased adult body weight and plasma IGF-1 levels, and a nearly 50% reduction in lifespan [44,45,46,47].
PI3K/Akt/mTOR signaling is a key regulator of glycolytic muscle homeostasis, hypertrophic growth, and metabolism through its activation of mTOR-dependent anabolic processes while simultaneously inhibiting both autophagy and Foxo1/3 catabolic processes [48,49]. Skeletal muscle-specific transgenic overexpression of Akt selectively promotes muscle hypertrophy [50]. Glycolytic muscle specific transgenic overexpression of Akt similarly induces glycolytic muscle hypertrophy, promoting weight loss and insulin sensitivity in obese [51] and aged mice [52]. Functional overload [53], IGF-1 stimulation [54,55], and skeletal muscle specific transgenic overexpression of IGF-1 [56] positively regulate PI3K/Akt/mTOR signaling in muscle and promote hypertrophic growth. The phospho-activation of Akt is initiated by a series of intracellular signaling events arising from receptor tyrosine kinases (RTKs) including the IGF-1 (IGF-1R) and insulin (Insr) receptors, and G-protein-coupled receptors (GPCR), which signal through scaffolding proteins (e.g., IRS1/2, Grb2/10) to activate PI3K. Active PI3K produces phosphatidylinositol (3,4,5)-trisphosphate (PIP3), leading to the phospho-activation of Akt at T308 by PDK1. Maximal activation of Akt requires phosphorylation at the S473 site, which is directly phosphorylated by mTORC2 [26,29]. Comprised of core components mTor, Rictor, SIN1, and mLST8, mTORC2 functions primarily as an effector of insulin/PI3K activity [57]. Though the regulation of mTORC2 is uncertain, a growing body of evidence would suggest that mTORC2 activity is positively modulated through its association with ribosomes, the mitochondrial-associated ER membrane, and lysosomes [58,59,60,61,62,63,64,65,66]. Physiological regulation of Akt activation is achieved, in part, by the PIP3 phosphatase PTEN and members of inositol polyphosphate family of Type IA phosphatases (e.g., Inpp4a, Inpp5a) [57,67,68]. Though critical in limiting Akt activation, PTEN is now being recognized for its role in modulating insulin sensitivity and energy expenditure [69]; mice with chronic diabetes or insulin resistance have increased PTEN protein expression [70], while PTEN transgenic mice have decreased muscle mass coincident with increased energy expenditures [71].
Activated Akt acts directly on multiple targets to modulate cellular growth and metabolic processes (reviewed in [29]). Akt phosphorylates TSC2 to repress the GAP activity of the TSC1/TSC2 complex toward RHEB (Figure 1), maintaining RHEB in the GTP-bound state (RHEB•GTP) to activate mTORC1 [72,73,74]. In cancer cells resistant to PI3K inhibitors, SGK1 similarly phosphorylates TSC2 (Figure 1) to activate mTORC1 [75] in response to direct phosphorylation at S422 by mTORC2 [76,77] or PDK1 [78,79]. In addition, direct phosphorylation of PRAS40 at T246 by Akt blocks its inhibition of mTORC1 [80,81,82]. Although PRAS40 can inhibit RHEB•GTP activation of mTORC1 [80], insulin signaling in Tsc2 null MEFs expressing a phospho-deficient form of TSC2 failed to induce mTORC1 signaling despite the phosphorylation of PRAS40 at T246 [83], suggesting that TSC-RHEB regulation of mTORC1 activation predominates. GSK3 is a constitutively active Ser/Thr protein kinase with a large set of functionally diverse direct targets, the majority of which are functionally inhibited or degraded following GSK3-directred phosphorylation [84,85]. FoxO transcription factors are associated with longevity in humans [86], in part, through modulating the expression of genes involved in apoptosis, glucose metabolism, cell cycle, DNA damage repair, and proteolysis [87]. FoxO proteins are exported from the nucleus and sequestered in the cytoplasm when phosphorylated at their N-terminus and NLS sequences (T24 and S256 on FoxO1) [88]. Akt phosphorylates both GSK3 [89,90] and FoxO transcription factors [91,92] to inhibit their respective kinase and transactivating activities.
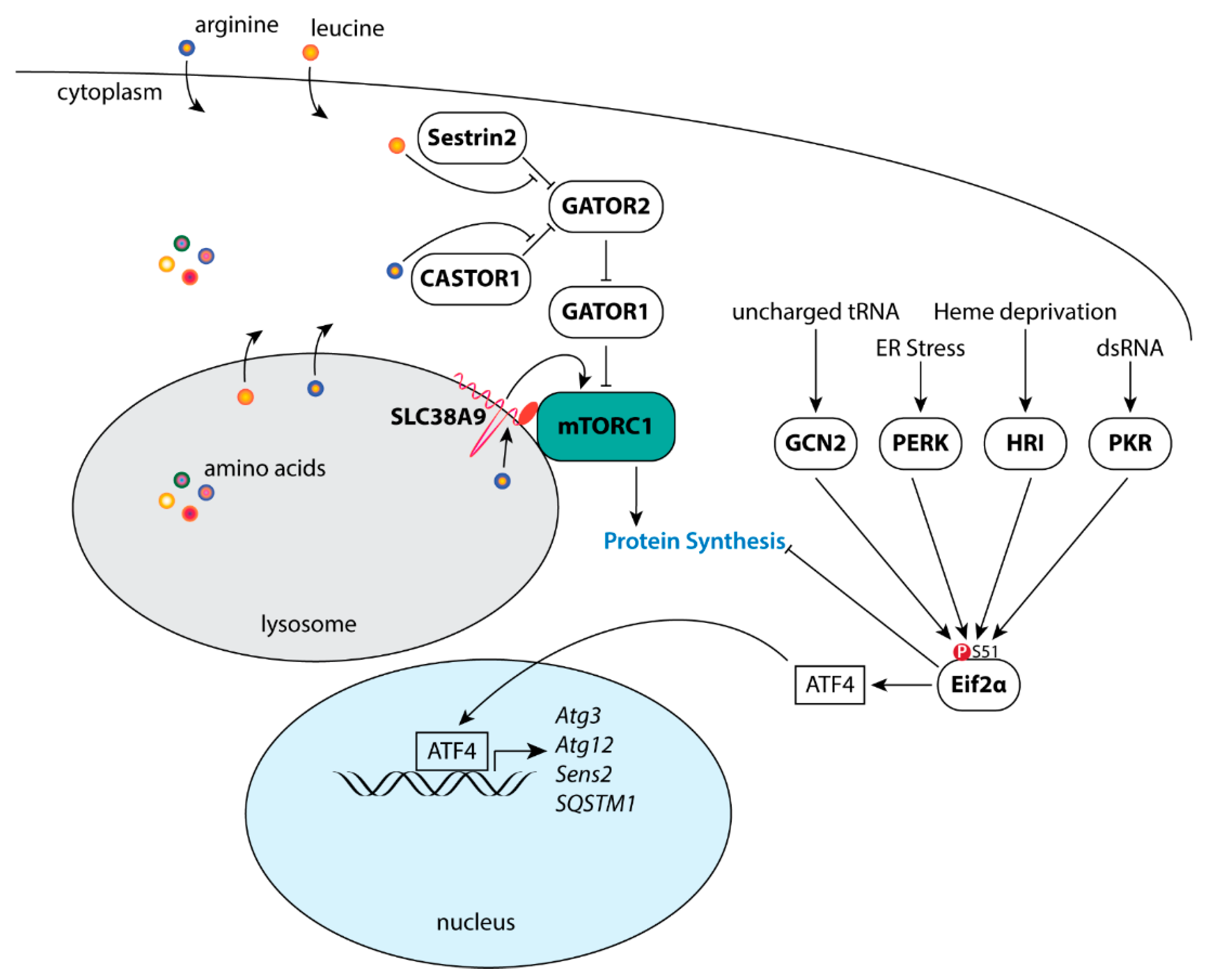
The mTOR-containing complex 1 (mTORC1) is a major effector of activated Akt, stimulating the synthesis of proteins, lipids, and nucleotides, while inhibiting protein degradation pathways [57]. Recruitment of the small ribosomal subunit to mRNA is dependent upon binding of the mRNA 5′ cap structure by the eukaryotic translation initiation factor 4F (eIF4F) complex. Comprised of the initiation factors eIF4E, eIF4G, and eiF4A, the eIF4F complex is critical for cap-dependent protein synthesis [93,94]. mTORC1 directly phosphorylates the eIF4E inhibitor 4E-BP1 at T37/46 to promote assembly of the eIF4F complex on the 5′ cap of mRNA [57,95,96]. In addition, mTORC1 phosphorylates S6K1 at T389 [97,98], activating its kinase activity toward the 40S ribosomal protein S6 (rpS6) [99], eIF4B [100], and the eIF4B inhibitor PDCD4 [101] to stimulate protein synthesis. Phosphorylation of eIF4B at S422 by S6K1/2 stimulates eIF4F activity by potentially promoting the ATPase, RNA-binding, and RNA-helicase activities of eIF4A [93,100,102]. Akt/mTORC1 signaling promotes de novo lipid synthesis through promoting the nuclear accumulation of SREBP to initiate the transcription of metabolic genes involved in fatty acid synthesis [103,104]. Further, pyrimidine synthesis is stimulated through S6K1 phosphorylation of CAD [105], while mTORC1-dependent purine synthesis is through activation of the mitochondrial tetrahydrofolate cycle, in part, through induction of MTHDF2 by eIF2α-independent activation of ATF4 [106]. In response to cellular stress (e.g., amino acid insufficiency, unfolded protein response of the endoplasmic reticulum) Eif2α is phosphorylated at S51 (Figure 1) to both inhibit cap-dependent mRNA translation and to initiate the transcription of stress response genes including FGF21, SQSTM1, ATG3, ATG12, and Sestrin2 (Sesn2) via ATF4 [107,108,109]. Lastly, autophagy is an essential process for the maintenance of cellular homeostasis that may be further induced in response to cellular stress to reduce organelles and macromolecules into amino acids, nucleosides, fatty acids and sugars in support of cellular growth and survival [110]. Under growth conditions, active mTORC1 negatively regulates autophagy (Figure 1) through its direct phosphorylation of ULK1 at S757 [111] and Atg13 at S258 [112,113].
Nutrient and energy availability is a critical regulator of mTORC1. Increases and decreases in amino acids leucine and arginine positively and negatively modulate mTORC1 kinase activity via CASTOR1, Sestrin2, and SLC38A9 [114,115,116,117,118], thus providing a critical feedback mechanism to modulate mTORC1 activity in response to nutrient availability. Similarly, under low ATP conditions, AMPK may negatively modulate mTORC1 kinase activity (Figure 1) through phosphorylation of the mTORC1-specific constituent scaffolding protein Raptor [119] or through the phospho-activation of TSC2 [120,121]. Further, AMPK phosphorylates mTOR at S1261 within mTORC2, while AICAR-induced AMPK activation both increases and decreases mTOR autophosphorylation at S2481 when associated with Rictor and Raptor, respectively [62]. Although the mTOR S1261 phosphorylation was found to be dispensable for the AMPK-dependent increase in mTORC2 catalytic activity, AMPK-mTORC2 signaling was found to promote cell survival following glucose withdrawal.
Multiple human studies have indicated significantly reduced rates of mixed, myofibrillar and/or mitochondrial protein synthesis in the muscles of elderly subjects [122,123,124,125,126,127,128,129]. However, other more recent human studies show little or no difference in basal muscle protein synthesis rates between the young and elderly [8,130,131,132,133,134,135,136]. While differences in physical activity level, genetic makeup, and nutritional input clearly contribute to these earlier observations, the previously reported >30% reductions in basal muscle protein synthesis rates [122,123,124,125,126,127,128,129] may not be reflective of normal physiological aging. Differences in muscle biopsy locations, analysis methods accounting for myofibrillar and total protein synthesis rates, and/or subject screening for average physical activity levels, nutritional intake, and overall health may similarly contribute. Additional studies with larger cohorts utilizing methods with improved sensitivity to protein synthesis while incorporating measures of protein degradation (e.g., proteosomal and autophagic flux) are warranted.
Resistance training increases muscle protein synthesis in both young and elderly men and women [122,124,126,128,137,138,139,140]. Exogenous and post-prandial amino acids increase muscle protein synthesis in both young and elderly men and women at baseline [8,133,141,142,143], while providing an additive effect post exercise [144]. However, the efficacy of both exercise and amino acids on stimulating muscle protein synthesis is blunted in the elderly [8,139,145], a phenomenon referred to as anabolic resistance. This occurs despite increased plasma concentrations of leucine in elderly subjects, as compared to young, following both exercise and ingestion of essential amino acids [8]. Further, elderly individuals, as compared to young controls, have blunted inductions of Akt phosphorylation at T308 and S473, S6K1 phosphorylation at T389, and 4E-BP1 phosphorylation at T37/46 after both exercise alone, and amino acid supplementation after exercise [145,146], strongly suggesting that PI3K/Akt/mTOR signaling dysregulation mechanistically contributes to anabolic resistance.
In vivo mechanistic studies in animal models add further support to this notion. Reductions in Akt phosphorylation at S473 and S6K1 phosphorylation at T389 are observed in the gastrocnemius of old mice as compared to the young controls [147]. This study further employed targeted transcriptomic and metabolomic analyses, revealing dysregulation of fatty acid and glucose metabolic processes in old mice. In contrast, significant increases in basal Akt phosphorylation at S473 [148] and S6K1 phosphorylation at T389 [149], are observed in the muscles of 33- and 24-month old rats, respectively. RpS6 phosphorylation at S240/244 is similarly increased in the 24-month old rats [149], indicating activation of a subset of anabolic signaling branchpoints. It should be noted that the increase in Akt S473 phosphorylation at 33-months was observed in the slow oxidative soleus, rather than fast glycolytic muscles predominantly examined for Akt signaling. Interestingly, the slow oxidative myofibers of the soleus, similar to fast glycolytic myofibers of the gastrocnemius of rats, positively modulate Akt phosphorylation at S473 following sciatic nerve stimulation [150] and 30 min of treadmill exercise [151]. Importantly, glycogen content in the soleus (65% reduction) was reduced to a greater extent than in the red (47% reduction) and white (23% reduction) gastrocnemius following treadmill exercise [151], suggesting that insulin-stimulated PI3K/Akt-regulated glucose uptake and glycogen storage is similarly activated in oxidative muscles in response to exercise. However, the extent to which Akt signaling modulates oxidative muscle metabolism through its influence on PGC-1α transcriptional activity in vivo [152], is uncertain and warrants future investigation.
The percentages of histologically abnormal myofibers, and myofibers staining positive for both phosphorylated rpS6 and active caspase-3 are increased in the muscles of both old mice and humans [153]. Counterintuitively, muscle-specific activation of mTORC1 (Tsc1 knockout) in mice induces progressive myofiber atrophy, phenocopying many of the histological features of aged muscles including increases in both myofiber vacuolation and the number of caspase-3 positive myofibers [153,154]. Similarly, pharmacological inhibition of mTORC1 reduced myofiber atrophy and the expression of both Atrogin-1 and MuRF1 in a mouse model of cancer cachexia [155]. In contrast, short-term activation of mTORC1 (TSC2 knockdown) specifically in skeletal muscle induced myofiber hypertrophy and greatly increased hypertrophy on recovery from transient denervation [156]. Consistent with these observations, muscle-specific inhibition of mTORC1 (Raptor knockout) in mice induced a leftward shift in the myofiber size distribution curve and histological features including centralized nuclei and vacuoles, and myofiber degeneration [157]. These apparently discordant observations with respect to myofiber size may more accurately be rationalized through the effects of Akt/mTORC1 signaling on metabolism, rather than simply the balance of protein synthesis and protein degradation. Short-term activation of Akt induces myofiber hypertrophy [51,52] in parallel with a metabolic shift from glycolysis and oxidative phosphorylation toward the accumulation of branched chain amino acids and enhancement of the pentose phosphate pathway (PPP) [158]. Importantly, PPP produces ribose 5-phosphate and NADPH which are critical for the production of nucleotides, fatty acids, sterols, non-essential amino acids, and cellular antioxidant defenses via reduced glutathione [159]. These observations would suggest that the hypertrophic effects of short-term Akt/mTORC1 signaling become deleterious when prolonged, prioritizing the synthesis of anabolic precursors rather than energy production, and leading to cellular and metabolic stress. Increased expression of pro-inflammatory cytokines (e.g., Ccl2, Ccl3, Il10) and metabolic stress indicators (e.g., FGF21, GDF3) in the hypertrophic muscles of Akt transgenic mice [158], and in atrophied muscles in which Akt is observed to be constitutively active [160], further support this notion.
Treatment with rapamycin both extends the lifespan of mice and fruit flies [161,162,163] and blocks resistance exercise-mediated increases in protein synthesis in humans [164,165]. Akt phosphorylation at both T308 and S473 are increased in muscle following a 16-h fast in 24- and 30-month old male mice with respect to 6-month old controls [166]. Male mice in this study also had significant increases in S6 phosphorylation at S240/244, whereas in contrast, aged female mice only showed an increase in Akt S473 phosphorylation without the increase in S6 phosphorylation after fasting [166]. Though suggestive of positive regulation of Akt via mTORC2 following a fast in aged mice, interpretation of these observations may be confounded by multiple variables including fasting duration, species, gender, systemic and muscle metabolic flux, as well as housing and dietary conditions throughout the lifespan of experimental animals. It is important to note that fasting in young healthy mice, in which systemic control of blood sugar and insulin is physiologically normal, induces the positive regulation of proteolytic processes through increased FoxO transcriptional activity [167] and AMPK induction of autophagy through phosphorylation of ULK1 at Serines 317, 555, and/or 777 (Figure 1) [111,168,169] while inhibiting protein synthesis [49,170]. Consistent with this, pharmacological inhibition of mTORC1 restored muscle histological and functional parameters in Tsc1 mutant mice [154,171] and in aged rats [149], in part, through restoration of autophagy.
3. Muscle Proteolytic Processes and Negative Regulation of Anabolic Processes
Aging and aging-related diseases are linked to impaired proteome integrity or proteostasis [172,173]. Acute and gradual increases in oxidative damage negatively impact protein stability and enzymatic function [174,175,176,177]; 40–50% of all proteins in old organisms are estimated to have some form of oxidative damage [178]. Similarly, the accumulation of misfolded proteins is known to contribute to the development of age-associated pathologies including Alzheimer’s disease, Huntington’s disease, and sporadic inclusion-body myopathy [172,173,179,180].
Cells have evolved multiple protein degradation processes to maintain proteome integrity or proteostasis in response to endogenous and exogenous stresses that negatively impact the protein functionality due to damage or misfolding. Approximately 60–80% of protein degradation in growing mammalian cells occurs through 26S proteasome catalysis [181,182,183,184]. Specificity is achieved through the selective polyubiquitylation of substrates by specific E3 ubiquitin ligases. While the proteasome can degrade some non-ubiquitinated proteins, ubiquitination may function independently of the proteasome by selectively directing proteins (typically membrane proteins) to the lysosome for proteolysis [184]. Similarly, autophagy reduces organelles and macromolecules into amino acids, nucleosides, fatty acids, and sugars in support of cellular growth and survival [110]. The core autophagy-related proteins essential for formation of the autophagosome formation and delivery of autophagic cargo to the lysosome may be grouped by functionally: (1) initiation and phagophore nucleation proteins ULK1, ULK2, ATG1, FIP200, ATG13, VPS34, ATG9, Beclin, (2) phagophore expansion proteins ATG3, ATG4, ATG7, ATG9, ATG10, ATG12, (3) cargo sequestration and membrane sealing proteins ubiquitin, cardiolipin, p62/SQSTM1, PE-conjugated LC3s and GABARAP, and (4) autophagosome maturation and lysosome fusion proteins ATG4, ATG14, VCP, and PE-conjugated LC3s and GABARAP [185,186].
In humans and animal models, these proteolytic processes become dysregulated with pathologies associated with muscle atrophy including cancer cachexia, chronic renal failure, diabetes, denervation, and age [187,188,189]. Atrophying muscles in old [190] and in young thyroidectomized and tumor bearing rats [191] have increased levels of protein polyubiquitylation. Consistent with the experimental models of disuse muscle atrophy in mice [192], increased levels of protein polyubiquitylation are observed in the muscles of young human volunteers following an extended 20-day bed rest [193]. Gene expression analyses during the early stages of muscle wasting due to denervation, disuse, diabetes, cachexia, and fasting have identified a common set of genes, referred to as atrogenes, whose expression is coordinately induced or repressed under conditions of muscle atrophy or growth. Atrogenes whose expression is increased in atrophying muscles include ubiquitin, core components of the 26S proteosome, eIF4E inhibitor 4E-BP1 (Eif4ebp1), transcription factors Atf4, Foxo1, and Foxo3 [194,195], the muscle specific E3 ubiquitin ligases MurF1 (Trim63) and Atrogin-1/MAFbx (Fbxo32) [196,197], and autophagosome components LC3 (Map1lc3a) and Gabarap [194,198,199]. Importantly, animal models indicate that both insufficient [200,201,202,203] and excessive [198,199,204] proteolysis, both proteasomal and autophagic, negatively impact the muscle mass and functionality, suggesting an inflection point between a level of proteolysis that supports muscle homeostasis and that which promotes muscle wasting.
Autophagy and ATF4 are both positively regulated in response to cellular stress (Figure 2) and major effectors of the integrated stress response (ISR). The ISR is a multifaceted signaling pathway that concurrently represses cap-dependent protein synthesis via Eif2α while inducing the translation of select mRNAs which contain short inhibitory upstream open reading frames (uORFs) in their 5′-untranslated regions including ATF4, DDIT3 (CHOP), and PPP1R15A (GADD34) [205]. The Eif2α kinases PERK, PKR, and HRI converge to phosphorylate Eif2α at S51 in response to the unfolded protein response of the endoplasmic reticulum (UPRER) [206,207], dsRNA [208,209], and heme deprivation [210,211], respectively. In addition, amino acid sensing plays a key role in both activating the ISR as well as inhibiting mTORC1 (Figure 2). Reduced levels of amino acids lead to both an increase in the levels of uncharged tRNAs thereby activating GCN2 kinase activity toward Eif2α [212], and a concurrent reduction in mTORC1 activity via Sestrin2 [115], CASTOR1 [213] and SLC38A9 [214,215,216].
FoxO transcription factors are positive regulators of Atrogin-1, MuRF1, LC3, GABARAPs, ULK2, ATG12, and Beclin [198,199,217,218], thus indicating their central role in promoting proteolytic processes. Transgenic overexpression of Foxo1 specifically in skeletal muscle results in mice with decreases in muscle mass, functionality, and sarcomeric protein expression [219], while muscle specific Foxo1 knockout mice were largely protected from nephrectomy and dexamethasone-induced muscle atrophy [220]. Muscle-specific genetic deletion of all three muscle Foxo transcription factors (Foxo1, Foxo3, and Foxo4) abolished both fasting [221] and insulin-deficient diabetes [222] induced muscle loss and weakness. Consistent with these observations, heterozygous deletion of either Atrogin-1 or MuRF1 results in mice that are partially protected from denervation-associated muscle atrophy [196]. Similarly, overexpression of a constitutively active form of FoxO3 in tibialis anterior muscles markedly increased the autophagosome formation and myofiber atrophy while siRNAs targeting Foxo3 and overexpression of a dominant negative form of FoxO3 blocked fasting-induced autophagy [198].
Proteins may also be cleaved by members of the caspase family of proteases which are known to play key roles in regulating apoptosis, necrosis, and inflammation [223,224]. Perhaps best known for its role in the activation of IL-1β [224], caspase-1 enzymatic activity is observed to be significantly increased in the muscles of 24-month old mice as compared to 10-month old controls [225]. Interestingly, genetic deletion of Nlrp3 blunted both the age-associated changes in muscle mass and caspase-1 activity, respectively [225], thus demonstrating a mechanistic link between the NLRP3 inflammasome and age-related muscle atrophy. The muscles of 24-month old rats show no statistical differences in either cytosolic cytochrome c levels or cytosolic caspase-3 activity from 6-month old controls [226], suggesting that mitochondrial-mediated caspase activation is not present at this timepoint. Further, unlike in liver homogenates, cytochrome c stimulation of muscle homogenates from 6- and 24-month old rats was unable to increase caspase-3 enzymatic activity, suggesting that additional factors may be necessary for caspase-3 activation in muscle. Acute insulinopenia stimulates non-lysosomal, ATP-, and proteasome-dependent proteolysis in rats [227]. Subsequent studies identified caspase-3 as the protease likely responsible for proteolyzing actomyosin into the 14-kDa actin fragments observed in the atrophying muscles of diabetic rats [228]. Subsequent studies identify caspase-3 as the protease responsible for proteolyzing actomyosin into the 14-kDa actin fragments observed in the atrophying muscles of diabetic rats [228] and following denervation injury in mice [229]. Importantly, the enzymatic activities of caspases-3, -6, -8, and -9 were found to be significantly increased in the gastrocnemius of cachexic (MAC16 tumor-bearing) as compared to non-cachexic (MAC13 tumor-bearing) mice [230]. Collectively, these studies suggest that increased caspase activity may play a causal role in the muscle atrophy associated with systemic metabolic dysfunction.
Sporadic inclusion body myopathy (sIBM) is the most common degenerative muscle disease in patients aged 50 years or more, and is often referred to as sporadic inclusion-body myositis due to high levels of immune cells, including cytotoxic CD8+ T cells, in the endomysium [231]. sIBM patients as a group respond poorly to anti-dysimmune therapies [232] suggesting that immune system dysfunction is likely not causal for sIBM, but rather exacerbates sIBM severity and/or progression. Muscle degeneration in sIBM is characterized by vacuolization and intra-myofiber accumulation of misfolded and polyubiquitinated protein aggregates including amyloid-β (Aβ), SQSTM1, LC3, TDP-43, HSP70, and proteasomal subunits [179,233,234,235,236,237]. In addition, biochemical analyses reveal an increase in LC3-II and SQSTM1 expression along with a decrease in the enzymatic activity of lysosomal enzymes Cathespin B and D despite their increased expression [236]. However, though the increase in LC3-II is indicative of mature autophagosomes, the increase in SQSTM1 would rather suggest a decrease in autophagic flux [238]. Consistent with these observations, transgenic mice overexpressing R155H or A232E VCP mutants commonly observed in humans with IBM associated with Paget’s disease recapitulate the progressive functional and morphological deficits observed in sIBM patients [239]. Treatment of VCP A232E transgenic mice with arimoclomol restored grip strength and maximal EDL tetanic force to near WT levels and markedly reduced ubiquitin and TDP-43 aggregates [240], indicating that dysregulated proteolysis plays a significant role in the pathogenesis of sIBM.
4. Unexpected Links
RNA-binding proteins (RBPs) play essential roles in nearly every aspect of cell biology through their direct influence on post-transcriptional gene expression [241,242,243], translational control [244], RNA quality and stability [245], and RNA splicing [246]. A census of human RBPs revealed that ~8% of protein coding genes are directly involved in RNA metabolism (the collective events in the life cycle of RNA molecules including synthesis, secondary structure modulation, base modification/editing, processing, and degradation), either through direct interaction or as essential components of ribonucleoprotein (RNP) complexes [247]. Importantly, alterations in RNA metabolic processes are increasingly being recognized for its contributions to physiological aging and age-associated disease. The roles of RBPs in skeletal muscle typically have been studied in the context of developmental and/or disease processes. Identifying mechanistic links between the activities of RBPs and the cellular processes modulating skeletal muscle physiology in the context of aging is an ongoing pursuit. Here, we will highlight recently identified, and potentially new links, between the activity of RBPs, biological aging, and skeletal muscle.
Altered post-transcriptional gene expression regulation, in part through changes in miRNA expression, play causal roles in the cellular and tissue morphological changes in the cardiovascular systems of the elderly [248,249,250]. SNPs in A-to-I RNA editing genes ADARB1 and ADARB2 are associated with longevity in both humans and Caenorhabditis elegans [251], and alterations in the editing of the transcriptome is implicated in the etiology of disease [252,253]. In mice, genetic deletion of Adar induced lethality around ED11.5 due to severe defects in hematopoiesis and disintegration of the liver [254]. Similarly, mice in which Adarb2 is genetically deleted develop normally, but die between P0 and P20, becoming progressively seizure prone by P12 [255]. In addition, proteome analyses of 7- and 30-month old rat gastrocnemius muscles indicates a significant increase in the expression of the C-to-U RNA editing enzyme APOBEC2 [256], suggesting that RNA editing in general, and specific subsets in particular (e.g., A-to-I, C-to-U) are temporally regulated in a cell type and context-dependent manner. Interestingly, such regulation may be in response to IIS signaling. ADAR2 expression in pancreatic islets, as well as ADAR2-targeted editing of GluR-B RNA, is positively regulated in obese insulin resistant mice with hyperinsulinemia [257]. A recent study identified ADAR1 T738 (Adar) and ADAR2 T553 (Adarb2) as being direct targets of Akt kinase activity in cultured human cell lines [258]. Akt inhibition increased, whereas phospho-mimetic mutants of the ADAR1 and ADAR2 Akt sites reduced the RNA editing of select transcripts including NEIL1, CCNI, AZIN1, and CYFIP2. Interestingly, millions of RNA editing sites have been identified in human cells [259,260]. However, the vast majority of editing sites in humans and primates are observed in the ubiquitous inverted repeat Alu elements largely found in noncoding regions of the genome [260]. Further, and in-depth cDNA sequencing analysis of the brain of a 67-year old male concluded that less than 119 of the transcript sequence variants identified from 541,777 nt of exon sequences analyzed are likely due to post-transcriptional RNA editing [261]. Although these observations would suggest that RNA editing plays a negligible role in introducing post-transcriptional amino acid substitutions in humans, additional studies in young and old individuals are necessary to determine whether RNA editing plays an extensive role in biological aging.
Alterations in RNA processing steps including splicing, poly-adenylation, and 5′ capping) are associated with both physiological aging and age-associated disease [20,262,263,264,265]. It is estimated that >95% of human multi-exon genes are subject to alternative splicing [266]. Brain, heart, and skeletal muscle have the highest levels of evolutionarily conserved alternatively spliced transcripts [267], and age-associated changes in splicing are observed in the transcriptomes of muscles from aged humans [20,268,269] and mice [270]. Importantly, changes in alternative splicing may drive cellular dysfunction through changes in the binding properties, enzymatic activities, and intracellular localization of a large percentage of the proteome [20,262,271], and is implicated as a causal mechanism of cellular aging through its impact on both metabolic processes and DNA repair [272]. In mammals, ~40 proteins and 5 small nuclear RNAs form the core spliceosomal complex [273,274], which is dynamically regulated by the activities of multiple RBPs, including members of the SR protein [275] and heterogeneous nuclear ribonucleoprotein (hnRNP) [276] families. Individual transcript splicing is modulated, in part, by cis-acting elements within the nascent transcript (e.g., exonic splicing enhancers/silencers—ESE/ESS, intronic splicing enhancers/silencers—ISE/ISS) and secondary structure [277]. Importantly, tissue-specific and ubiquitously expressed RBPs cooperatively interact with the regulatory elements both within the nascent transcript and of the spliceosome for context- and tissue-specific mRNA splicing [246,278,279].
Genetic mutations in the splicing factor gene RBFOX1 have been observed in patients with neurological disorders [280] and in an autistic individual with muscle weakness [281]. Mice in which Rbfox1 has been specifically deleted in skeletal muscle have decreases in muscle mass and maximal force generation [282], thus establishing a role for Rbfox1 in muscle development and physiology. In addition, mice in which both Rbfox1 and Rbfox2 are specifically deleted in adult mice, experienced rapid losses of muscle mass, myofiber cross sectional area, and strength, due in part, to reductions in autophagy and the aberrant expression of calpain-3 [283]. Further, siRNAs targeting Rbfox1 and/or Rbfox2 significantly impaired myogenesis in the C2C12 model system through its direct role in promoting the muscle-specific splicing of Mef2d [284]. Collectively, these studies establish Rbfox1/2 as essential for muscle developmental processes and the maintenance of muscle mass and strength in adulthood. Whether changes in the expression or activity of these factors contribute to age-associated muscle atrophy is uncertain.
Amyotrophic lateral sclerosis (ALS) is a progressive age-related neurodegenerative disease in which motor neurons are selectively degenerated. Both reductions in nuclear/cytoplasmic ratio of the splicing factor SFPQ, and the aberrant retention of SFPQ introns, are observed in motor neurons that were differentiated from ALS patient-derived iPSCs [285], thus suggesting a causal role for SFPQ in the pathogenesis of ALS. Neuron-specific deletion of Sfpq in mice led to impaired transcriptional elongation, an effect most pronounced on long genes (>100 kb), and gross developmental abnormalities in the brain [286]. Skeletal muscle-specific deletion of Sfpq similarly induced long gene transcriptopathy and premature death starting around P30 [287]. Between P14 and P30, these mice exhibited significant skeletal muscle growth defects, attributable to the combination of an increase in the percentage of oxidative myofibers, accumulation of excess glycogen, and a decrease in the abundance of OXPHOS complexes I, II, and IV. Although reductions in mitochondrial oxidative capacity are observed in aged and sarcopenic muscles [288,289,290], it is unknown whether dysregulation of SFPQ expression or activity plays a causal role.
A rare cohort of patients have been identified in which heterozygous mutations in HNRNPU are associated with intellectual disability and muscle weakness [291,292,293,294]. hnRNP-U is a multifunctional RBP, directly involved in regulating alternative splicing [295,296,297], genome architecture [298], X-chromosome inactivation [299], and is known to promote the DNA base repair through its interaction with NEIL1 [300]. In mice, hnRNP-U protein expression decreases with advancing age in both the heart [301] and skeletal muscle [160], suggesting that loss of hnRNP-U may contribute to age-associated pathologies in these tissues. Mice in which Hnrnpu is deleted in striated muscles (Ckm-Cre) die around P14 due to a dilated cardiomyopathy (DCM) phenotype, whereas mice in which Hnrnpu is deleted specifically within cardiomyocytes (Myh6-Cre) die around P10 due to a similar DCM phenotype [301]. Mice in which Hnrnpu is deleted specifically in skeletal muscle (ACTA1-Cre) are born phenotypically normal, but by 3-months of age develop a myopathy-like phenotype characterized by selective muscle atrophy of glycolytic muscles and histopathological observations of central nuclei, myofiber degeneration, intracellular inclusions, and extensive fibrosis [160]. Changes in the expression and splicing of genes are observed in both cardiac (Ckm-Cre) and skeletal muscles (ACTA1-Cre). Interestingly, the atrophied skeletal muscles (ACTA1-Cre) exhibit signs of metabolic stress. Akt is observed to be constitutive active with its phosphorylation at S473 being desensitized to IGF-1 stimulation, SQSTM1 and Ulk1 phosphorylation at S757 are significantly increased, and multiple genes involved in the transport/import and biosynthesis of amino acids, as well as the repression of oxidative metabolism are significantly reduced in expression [160]. Though high levels of Akt S473 [149] and S6K1 T389 [149] phosphorylation are similarly observed in the atrophied muscles of aged rats, it is unclear whether this is a compensatory effect, or a mechanistic contributor, likely only at specific stages of muscle atrophy.
Splicing of the insulin receptor gene INSR is regulated in a cell-type specific context that is dependent upon the development stage or disease condition. Alternative splicing of exon 11 gives rise to protein isoforms INSR-A (exclusion of exon 11) and INSR-B (inclusion of exon 11). INRS-A, which is prevalent during fetal development and in the nervous system, preferentially binds IGF-2 over IGF-1 and insulin, while INRS-B, which is prevalent in adulthood and highly expressed in adipose tissue, liver and skeletal muscle, is more sensitive to insulin [302,303,304]. The splicing of INSR is regulated by multiple RBPs including CELF1, SRSF3, MBNL1, and hnRNPs A1, F, and H [305,306,307,308]. Patients with myotonic dystrophy (DM) frequently acquire muscle insulin resistance. Expansion of CTG (DM type 1) and CCTG (DM type 2) repeats in DMPK [309] and ZNF9 [310], respectively, leads to the dysregulation of alternative splicing through MBNL1 loss-of-function and CELF1 gain-of-function [311]. Molecular analyses have identified hnRNP-H, MBNL1, and CELF1 as modulating the increase in the INSR-A:INSR-B ratio observed in DM patients [308,312,313,314]. Although insulin resistance in skeletal muscle is the primary defect in type 2 diabetes [315], and dysregulation of INSR splicing occurs in the insulin target tissues of patients with insulin resistance [312,313], the role of INSR splicing in the pathogenesis of type 2 diabetes is unclear [303]. Conflicting observations in which the transcript ratio of INSR-A:INSR-B splice variants is either decreased [316] or unchanged [317,318] are reported. Similarly, it has been reported that the INSR-A transcript is exclusively expressed in non-diabetics with the induction of the INSR-B transcript observed in the muscles of type 2 diabetics [319]. To the best of our knowledge, age-associated changes in muscle INSR protein isoform expression, and whether such isoform expression changes are correlated with the development of type 2 diabetes in the elderly is unknown.
Treatment of Ewing sarcoma cells with the ATP-competitive PI3K and mTOR inhibitor Dactolisib induced significant reductions in both cell growth and proliferation [320]. Genes for RBPs with known roles in splicing modulation including FUS, HNRNPM, SFPQ, and SRSF2 were significantly increased, while coimmunoprecipitation and cosedimentation of hnRNP-M with core spliceosomal proteins were significantly enhanced following treatment. Importantly, siRNA knockdown of HNRNPM blocked a subset of Dactolisib induced changes in cassette exon splicing for exons containing proximal hnRNP-M consensus motifs [320]. Interestingly, hnRNP-M expression is positively corelated with SGK1 phosphorylation, and its overexpression rescues the impaired differentiation of C2C12 myoblasts treated with siRNA targeting Rictor [321]. Though hnRNP-M physically interacts with Rictor, and siRNAs targeting either Rictor or HNRNPM repressed insulin-stimulated Akt S473 and SGK1 S422 phosphorylation to a similar degree, the molecular mechanism underlying these observations is unknown. hnRNP-E2 (PCBP2) was identified in a two-hybrid screen of a human bone marrow cDNA library with SIN1 (MAPKAP1), while siRNAs targeting either SIN1 (MAPKAP1) or hnRNP-E2 (PCBP2) similarly potentiate an increase in the percentage of apoptotic cells treated with either H2O2 or TNFα [322]. Though endogenous SIN1 and hnRNP-E2 coimmunoprecipitated in cell lysates and combined siRNA treatments had an additive effect on both H2O2 and TNFα induced apoptosis, suggesting their participation in a common signaling pathway, the molecular mechanism, presumably activating mTORC2, is unknown. SR protein SF2/ASF (SFRS1) positively modulates protein translation through an Akt-independent mechanism [323]. MEF and NIH 3T3 cells overexpressing SF2/ASF had significantly increased S6K1 and 4E-BP1 phosphorylation without increases is Akt phosphorylation at S473. Interestingly, SF2/ASF physically binds to both mTOR and the catalytic subunit of the PP2A phosphatase [324] suggesting that it may facilitate mTORC1-mediated phosphorylation of 4E-BP1 by either recruiting it to the mRNP complex via direct binding of mRNA ESE or perhaps by indirect inhibition of PP2A thereby biasing the phosphorylation-dephosphorylation kinetics of 4E-BP1 toward phosphorylation. Intriguingly, these observations suggest that RBPs involved in nuclear-cytoplasmic shuttling of RNA may positively regulate mTOR-dependent cell survival and growth processes, perhaps as part of a feed forward mechanism to insure selective translation of cell survival and growth transcriptional programs.
5. Conclusions
Worldwide, the number of individuals aged 65 years or older is expected to more than double by the year 2050 [325]. In the U.S. alone, it is estimated that approximately 45% of adults greater than 60 years of age have some degree of sarcopenia [326], contributing to the impaired ability to perform activities of daily living and the loss of independence in the elderly population. In contrast with other well-studied diseases, and despite the urgent need by an aging population, muscle atrophy and weakness are frequently considered signs of “normal aging.” Though it is managed by a regimen of resistance training and increased protein intake, only a subset of sarcopenic individuals see improvements in muscle mass, strength, and functionality [11,327]. In the nearly 30 years since the identification of mTOR [328,329,330], the complex and often interrelated signaling pathways regulating skeletal muscle mass, strength, and functionality have begun to be more clearly defined. New mechanisms and linkages continue to be discovered, defining new regulatory circuits while further refining the established pathways. Clearly, there has been much exciting progress in our understanding of these pathways and their contribution to muscle wasting disorders, but much remains to be done.
Author Contributions
Writing—original draft preparation, E.-J.L. and R.L.N.; writing—review and editing, E.-J.L. and R.L.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. The APC was funded by Brigham and Women’s Hospital Department of Orthopaedic Surgery.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Julia Charles for helpful discussions and feedback.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Abbreviation | Full name |
| Aβ | amyloid-β |
| ADARB1 | adenosine deaminase RNA specific B1 |
| ADARB2 | adenosine deaminase RNA specific B2 |
| ADAR | adenosine deaminase RNA specific |
| APOBEC2 | apolipoprotein B mRNA editing enzyme catalytic subunit 2 |
| CAD | carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase |
| FoxO1 | Forkhead boxO transcription factor 1 |
| FoxO3 | Forkhead boxO transcription factor 3 |
| GAP | GTPase activating protein |
| GCN2 | General control nonderepressible 2 kinase |
| GH | Growth hormone |
| GPCR | G-protein-coupled receptor |
| GSK3 | glycogen synthase kinase 3 |
| HRI | Heme-regulated inhibitor kinase |
| IGF-1 | Insulin like growth factor 1 |
| IIS | Insulin/IGF-1 signaling |
| miRNA | microRNA |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase 2 |
| mTORC1 | mTOR containing complex 1 |
| mTORC2 | mTOR containing complex 2 |
| PERK | PKR-like ER kinase |
| PI3K | phosphoinositide 3-kinase |
| PKR | Protein kinase RNA-activated |
| PRAS40 | proline-rich AKT substrate of 40 kDa |
| RHEB | Ras homolog enriched in brain |
| rpS6 | ribosomal protein S6 |
| RTK | Receptor tyrosine kinases |
| S6K1 | ribosomal protein S6 kinase 1 |
| sIBM | sporadic inclusion body myopathy/myositis |
| SNP | single nucleotide polymorphism |
| ULK1 | unc-51 like autophagy activating kinase 1 |
| ULK2 | unc-51 like autophagy activating kinase 2 |
References
- U.S. Cancer Statistics Working Group. US Cancer Statistics: 1999–2009 Incidence and Mortality Web-Based Report; USDHHS, CDC: Atlanta, GA, USA, 2013.
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. The State of Aging and Health in America 2013; Centers for Disease Control and Prevention, US Dept of Health and Human Services: Washington, DC, USA, 2013.
- Heron, M. Deaths: Leading causes for 2010. In National Vital Statistics Reports: From the Centers for Disease Control and Prevention; National Center for Health Statistics, National Vital Statistics System: Hyattsville, ML, USA, 2013; Volume 62, pp. 1–97. [Google Scholar]
- Caspersen, C.J.; Thomas, G.D.; Boseman, L.A.; Beckles, G.L.; Albright, A.L. Aging, diabetes, and the public health system in the United States. Am. J. Public Health 2012, 102, 1482–1497. [Google Scholar] [CrossRef]
- Facchini, F.S.; Hua, N.; Abbasi, F.; Reaven, G.M. Insulin resistance as a predictor of age-related diseases. J. Clin. Endocrinol. Metab. 2001, 86, 3574–3578. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, B.B.; Fujita, S.; Wolfe, R.R.; Mittendorfer, B.; Roy, M.; Rowe, V.L.; Volpi, E. Insulin resistance of muscle protein metabolism in aging. FASEB J. 2006, 20, 768–769. [Google Scholar] [CrossRef]
- Cuthbertson, D.; Smith, K.; Babraj, J.; Leese, G.; Waddell, T.; Atherton, P.; Wackerhage, H.; Taylor, P.M.; Rennie, M.J. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. 2005, 19, 422–424. [Google Scholar] [CrossRef]
- Thompson, L.V. Age-related muscle dysfunction. Exp. Gerontol. 2009, 44, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Doherty, T.J. Invited review: Aging and sarcopenia. J. Appl. Physiol. (1985) 2003, 95, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Walston, J.D. Sarcopenia in older adults. Curr. Opin. Rheumatol. 2012, 24, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Keller, K.; Engelhardt, M. Strength and muscle mass loss with aging process. Age and strength loss. Muscles Ligaments Tendons J. 2013, 3, 346–350. [Google Scholar] [CrossRef]
- Breen, L.; Phillips, S.M. Skeletal muscle protein metabolism in the elderly: Interventions to counteract the ‘anabolic resistance’ of ageing. Nutr. Metab. 2011, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Burd, N.A.; Gorissen, S.H.; van Loon, L.J. Anabolic resistance of muscle protein synthesis with aging. Exerc. Sport Sci. Rev. 2013, 41, 169–173. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Walsh, K.; Arany, Z. Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E3–E10. [Google Scholar] [CrossRef] [Green Version]
- Lang, T.; Streeper, T.; Cawthon, P.; Baldwin, K.; Taaffe, D.R.; Harris, T.B. Sarcopenia: Etiology, clinical consequences, intervention, and assessment. Osteoporos. Int. 2010, 21, 543–559. [Google Scholar] [CrossRef] [Green Version]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Deschenes, M.R. Effects of aging on muscle fibre type and size. Sports Med. 2004, 34, 809–824. [Google Scholar] [CrossRef]
- Skelton, D.A.; Kennedy, J.; Rutherford, O.M. Explosive power and asymmetry in leg muscle function in frequent fallers and non-fallers aged over 65. Age Ageing 2002, 31, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harries, L.W.; Hernandez, D.; Henley, W.; Wood, A.R.; Holly, A.C.; Bradley-Smith, R.M.; Yaghootkar, H.; Dutta, A.; Murray, A.; Frayling, T.M.; et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 2011, 10, 868–878. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, H.Z.; Liu, D.P. The Four Layers of Aging. Cell Syst. 2015, 1, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, M.; Bonafe, M.; Franceschi, C.; Paolisso, G. Insulin/IGF-I-signaling pathway: An evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1064–E1071. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion blocks apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Brown-Borg, H.M. Hormonal control of aging in rodents: The somatotropic axis. Mol. Cell Endocrinol. 2009, 299, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Al-Regaiey, K.A.; Masternak, M.M.; Bonkowski, M.; Sun, L.; Bartke, A. Long-lived growth hormone receptor knockout mice: Interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction. Endocrinology 2005, 146, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Bartke, A.; Brown-Borg, H. Life extension in the dwarf mouse. Curr. Top. Dev. Biol. 2004, 63, 189–225. [Google Scholar] [CrossRef]
- Brown-Borg, H.M.; Borg, K.E.; Meliska, C.J.; Bartke, A. Dwarf mice and the ageing process. Nature 1996, 384, 33. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A.; Wright, J.C.; Mattison, J.A.; Ingram, D.K.; Miller, R.A.; Roth, G.S. Extending the lifespan of long-lived mice. Nature 2001, 414, 412. [Google Scholar] [CrossRef]
- Ikeno, Y.; Bronson, R.T.; Hubbard, G.B.; Lee, S.; Bartke, A. Delayed occurrence of fatal neoplastic diseases in ames dwarf mice: Correlation to extended longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Bokov, A.F.; Garg, N.; Ikeno, Y.; Thakur, S.; Musi, N.; DeFronzo, R.A.; Zhang, N.; Erickson, R.C.; Gelfond, J.; Hubbard, G.B.; et al. Does reduced IGF-1R signaling in Igf1r+/- mice alter aging? PLoS ONE 2011, 6, e26891. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gontier, G.; Chaker, Z.; Lacube, P.; Dupont, J.; Holzenberger, M. Longevity effect of IGF-1R(+/−) mutation depends on genetic background-specific receptor activation. Aging Cell 2014, 13, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Quipildor, G.F.; Tabrizian, T.; Novaj, A.; Guan, F.; Walters, R.O.; Delahaye, F.; Hubbard, G.B.; Ikeno, Y.; Ejima, K.; et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat. Commun. 2018, 9, 2394. [Google Scholar] [CrossRef] [Green Version]
- Berryman, D.E.; List, E.O.; Coschigano, K.T.; Behar, K.; Kim, J.K.; Kopchick, J.J. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm. IGF Res. 2004, 14, 309–318. [Google Scholar] [CrossRef]
- Bartke, A.; Chandrashekar, V.; Bailey, B.; Zaczek, D.; Turyn, D. Consequences of growth hormone (GH) overexpression and GH resistance. Neuropeptides 2002, 36, 201–208. [Google Scholar] [CrossRef] [PubMed]
- McGrane, M.M.; Yun, J.S.; Moorman, A.F.; Lamers, W.H.; Hendrick, G.K.; Arafah, B.M.; Park, E.A.; Wagner, T.E.; Hanson, R.W. Metabolic effects of developmental, tissue-, and cell-specific expression of a chimeric phosphoenolpyruvate carboxykinase (GTP)/bovine growth hormone gene in transgenic mice. J. Biol. Chem. 1990, 265, 22371–22379. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Brinster, R.L.; Hammer, R.E.; Trumbauer, M.E.; Rosenfeld, M.G.; Birnberg, N.C.; Evans, R.M. Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature 1982, 300, 611–615. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.M.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef] [Green Version]
- Izumiya, Y.; Hopkins, T.; Morris, C.; Sato, K.; Zeng, L.; Viereck, J.; Hamilton, J.A.; Ouchi, N.; LeBrasseur, N.K.; Walsh, K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008, 7, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Akasaki, Y.; Ouchi, N.; Izumiya, Y.; Bernardo, B.L.; Lebrasseur, N.K.; Walsh, K. Glycolytic fast-twitch muscle fiber restoration counters adverse age-related changes in body composition and metabolism. Aging Cell 2014, 13, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Takahashi, A.; Kureishi, Y.; Yang, J.; Luo, Z.; Guo, K.; Mukhopadhyay, D.; Ivashchenko, Y.; Branellec, D.; Walsh, K. Myogenic Akt signaling regulates blood vessel recruitment during myofiber growth. Mol. Cell Biol. 2002, 22, 4803–4814. [Google Scholar] [CrossRef] [Green Version]
- Musaro, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; Sweeney, H.L.; Rosenthal, N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Boulbes, D.R.; Shaiken, T.; Sarbassov dos, D. Endoplasmic reticulum is a main localization site of mTORC2. Biochem. Biophys. Res. Commun. 2011, 413, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.J.; Wu, C.C.; Kim, S.J.; Facchinetti, V.; Julien, L.A.; Finlan, M.; Roux, P.P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef] [Green Version]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Bonifacino, J.S. Lysosome Positioning Influences mTORC2 and AKT Signaling. Mol. Cell 2019, 75, 26–38.e23. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. PIP2 and PIP3: Complex roles at the cell surface. Cell 2000, 100, 603–606. [Google Scholar] [CrossRef] [Green Version]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.K.; Lopez-Knowles, E.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Molina, A.; Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 2013, 24, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Lee, I.H.; Wang, X.; Sheng, H.; Zhang, L.; Du, J.; Mitch, W.E. PTEN expression contributes to the regulation of muscle protein degradation in diabetes. Diabetes 2007, 56, 2449–2456. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Larrea, M.D.; Doughty, C.; Kwiatkowski, D.J.; Squillace, R.; Slingerland, J.M. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol. Cell 2008, 30, 701–711. [Google Scholar] [CrossRef]
- Kobayashi, T.; Cohen, P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 1999, 339 Pt 2, 319–328. [Google Scholar] [CrossRef]
- Biondi, R.M.; Kieloch, A.; Currie, R.A.; Deak, M.; Alessi, D.R. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001, 20, 4380–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J. Biol. Chem. 2003, 278, 10189–10194. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gan, L.; Pan, H.; Guo, S.; He, X.; Olson, S.T.; Mesecar, A.; Adam, S.; Unterman, T.G. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 2002, 277, 45276–45284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Vandenheede, J.R.; McDowell, H.E.; Hundal, H.S.; Cohen, P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: Evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem. J. 1994, 303 Pt 1, 21–26. [Google Scholar]
- Kops, G.J.; de Ruiter, N.D.; De Vries-Smits, A.M.; Powell, D.R.; Bos, J.L.; Burgering, B.M. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 1999, 398, 630–634. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Merrick, W.C. eIF4F: A retrospective. J. Biol. Chem. 2015, 290, 24091–24099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.; Blenis, J.; Pende, M.; Sonenberg, N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Rozen, F.; Edery, I.; Meerovitch, K.; Dever, T.E.; Merrick, W.C.; Sonenberg, N. Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol. Cell Biol. 1990, 10, 1134–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Palm, W.; Peng, M.; King, B.; Lindsten, T.; Li, M.O.; Koumenis, C.; Thompson, C.B. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 2015, 29, 2331–2336. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Yarasheski, K.E.; Zachwieja, J.J.; Bier, D.M. Acute effects of resistance exercise on muscle protein synthesis rate in young and elderly men and women. Am. J. Physiol. 1993, 265, E210–E214. [Google Scholar] [CrossRef]
- Yarasheski, K.E.; Welle, S.; Nair, K.S. Muscle protein synthesis in younger and older men. JAMA 2002, 287, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Welle, S.; Thornton, C.; Statt, M. Myofibrillar protein synthesis in young and old human subjects after three months of resistance training. Am. J. Physiol. 1995, 268, E422–E427. [Google Scholar] [CrossRef] [PubMed]
- Welle, S.; Thornton, C.; Jozefowicz, R.; Statt, M. Myofibrillar protein synthesis in young and old men. Am. J. Physiol. 1993, 264, E693–E698. [Google Scholar] [CrossRef]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Nair, K.S. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E92–E101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 15364–15369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasten, D.L.; Pak-Loduca, J.; Obert, K.A.; Yarasheski, K.E. Resistance exercise acutely increases MHC and mixed muscle protein synthesis rates in 78–84 and 23–32 yr olds. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E620–E626. [Google Scholar] [CrossRef] [PubMed]
- Balagopal, P.; Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am. J. Physiol. 1997, 273, E790–E800. [Google Scholar] [CrossRef] [PubMed]
- Volpi, E.; Sheffield-Moore, M.; Rasmussen, B.B.; Wolfe, R.R. Basal muscle amino acid kinetics and protein synthesis in healthy young and older men. JAMA 2001, 286, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Volpi, E.; Mittendorfer, B.; Wolf, S.E.; Wolfe, R.R. Oral amino acids stimulate muscle protein anabolism in the elderly despite higher first-pass splanchnic extraction. Am. J. Physiol. 1999, 277, E513–E520. [Google Scholar] [CrossRef]
- Volpi, E.; Mittendorfer, B.; Rasmussen, B.B.; Wolfe, R.R. The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J. Clin. Endocrinol. Metab. 2000, 85, 4481–4490. [Google Scholar] [CrossRef] [Green Version]
- Paddon-Jones, D.; Sheffield-Moore, M.; Zhang, X.J.; Volpi, E.; Wolf, S.E.; Aarsland, A.; Ferrando, A.A.; Wolfe, R.R. Amino acid ingestion improves muscle protein synthesis in the young and elderly. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E321–E328. [Google Scholar] [CrossRef]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. Am. J. Clin. Nutr. 2005, 82, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E381–E387. [Google Scholar] [CrossRef] [Green Version]
- Markofski, M.M.; Dickinson, J.M.; Drummond, M.J.; Fry, C.S.; Fujita, S.; Gundermann, D.M.; Glynn, E.L.; Jennings, K.; Paddon-Jones, D.; Reidy, P.T.; et al. Effect of age on basal muscle protein synthesis and mTORC1 signaling in a large cohort of young and older men and women. Exp. Gerontol. 2015, 65, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.J.; Churchward-Venne, T.A.; Parise, G.; Bellamy, L.; Baker, S.K.; Smith, K.; Atherton, P.J.; Phillips, S.M. Acute post-exercise myofibrillar protein synthesis is not correlated with resistance training-induced muscle hypertrophy in young men. PLoS ONE 2014, 9, e89431. [Google Scholar] [CrossRef]
- Yarasheski, K.E.; Pak-Loduca, J.; Hasten, D.L.; Obert, K.A.; Brown, M.B.; Sinacore, D.R. Resistance exercise training increases mixed muscle protein synthesis rate in frail women and men >/=76 yr old. Am. J. Physiol. 1999, 277, E118–E125. [Google Scholar] [CrossRef]
- Kumar, V.; Selby, A.; Rankin, D.; Patel, R.; Atherton, P.; Hildebrandt, W.; Williams, J.; Smith, K.; Seynnes, O.; Hiscock, N.; et al. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. J. Physiol. 2009, 587, 211–217. [Google Scholar] [CrossRef]
- Chesley, A.; MacDougall, J.D.; Tarnopolsky, M.A.; Atkinson, S.A.; Smith, K. Changes in human muscle protein synthesis after resistance exercise. J. Appl. Physiol. (1985) 1992, 73, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Volpi, E.; Ferrando, A.A.; Yeckel, C.W.; Tipton, K.D.; Wolfe, R.R. Exogenous amino acids stimulate net muscle protein synthesis in the elderly. J. Clin. Investig. 1998, 101, 2000–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpi, E.; Kobayashi, H.; Sheffield-Moore, M.; Mittendorfer, B.; Wolfe, R.R. Essential amino acids are primarily responsible for the amino acid stimulation of muscle protein anabolism in healthy elderly adults. Am. J. Clin. Nutr. 2003, 78, 250–258. [Google Scholar] [CrossRef]
- Luiking, Y.C.; Deutz, N.E.; Memelink, R.G.; Verlaan, S.; Wolfe, R.R. Postprandial muscle protein synthesis is higher after a high whey protein, leucine-enriched supplement than after a dairy-like product in healthy older people: A randomized controlled trial. Nutr. J. 2014, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Atherton, P.J.; Smith, K. Muscle protein synthesis in response to nutrition and exercise. J. Physiol. 2012, 590, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, C.S.; Drummond, M.J.; Glynn, E.L.; Dickinson, J.M.; Gundermann, D.M.; Timmerman, K.L.; Walker, D.K.; Dhanani, S.; Volpi, E.; Rasmussen, B.B. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle 2011, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Francaux, M.; Demeulder, B.; Naslain, D.; Fortin, R.; Lutz, O.; Caty, G.; Deldicque, L. Aging Reduces the Activation of the mTORC1 Pathway after Resistance Exercise and Protein Intake in Human Skeletal Muscle: Potential Role of REDD1 and Impaired Anabolic Sensitivity. Nutrients 2016, 8, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Argmann, C.; Houten, S.M.; Canto, C.; Jeninga, E.H.; Andreux, P.A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. The metabolic footprint of aging in mice. Sci. Rep. 2011, 1, 134. [Google Scholar] [CrossRef]
- Wu, M.; Katta, A.; Gadde, M.K.; Liu, H.; Kakarla, S.K.; Fannin, J.; Paturi, S.; Arvapalli, R.K.; Rice, K.M.; Wang, Y.; et al. Aging-associated dysfunction of Akt/protein kinase B: S-nitrosylation and acetaminophen intervention. PLoS ONE 2009, 4, e6430. [Google Scholar] [CrossRef] [PubMed]
- Joseph, G.A.; Wang, S.X.; Jacobs, C.E.; Zhou, W.; Kimble, G.C.; Tse, H.W.; Eash, J.K.; Shavlakadze, T.; Glass, D.J. Partial Inhibition of mTORC1 in Aged Rats Counteracts the Decline in Muscle Mass and Reverses Molecular Signaling Associated with Sarcopenia. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Hirshman, M.F.; Aschenbach, W.G.; Goodyear, L.J. Contraction regulation of Akt in rat skeletal muscle. J. Biol. Chem. 2002, 277, 11910–11917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Aschenbach, W.G.; Hirshman, M.F.; Goodyear, L.J. Akt signaling in skeletal muscle: Regulation by exercise and passive stretch. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1081–E1088. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Shrager, J.B.; Goldman, D. Rapamycin protects aging muscle. Aging 2019, 11, 5868–5870. [Google Scholar] [CrossRef]
- Castets, P.; Lin, S.; Rion, N.; Di Fulvio, S.; Romanino, K.; Guridi, M.; Frank, S.; Tintignac, L.A.; Sinnreich, M.; Ruegg, M.A. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013, 17, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S.; Handschin, C.; Tintignac, L.A.; Hall, M.N.; Ruegg, M.A. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet Muscle 2013, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Romanino, K.; Cloetta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.L.; Satomi, Y.; Walsh, K. RNA-seq and metabolomic analyses of Akt1-mediated muscle growth reveals regulation of regenerative pathways and changes in the muscle secretome. BMC Genom. 2017, 18, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, D.; Mason, B.D.; Baldino, K.; Li, B.; Lee, E.J.; Zhang, Y.; Chu, L.K.; El Raheb, S.; Sinha, I.; Neppl, R.L. Adult-Onset Myopathy with Constitutive Activation of Akt following the Loss of hnRNP-U. iScience 2020, 23, 101319. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Bitto, A.; Ito, T.K.; Pineda, V.V.; LeTexier, N.J.; Huang, H.Z.; Sutlief, E.; Tung, H.; Vizzini, N.; Chen, B.; Smith, K.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 2016, 5. [Google Scholar] [CrossRef]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Drummond, M.J.; Fry, C.S.; Glynn, E.L.; Dreyer, H.C.; Dhanani, S.; Timmerman, K.L.; Volpi, E.; Rasmussen, B.B. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J. Physiol. 2009, 587, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.M.; Fry, C.S.; Drummond, M.J.; Gundermann, D.M.; Walker, D.K.; Glynn, E.L.; Timmerman, K.L.; Dhanani, S.; Volpi, E.; Rasmussen, B.B. Mammalian target of rapamycin complex 1 activation is required for the stimulation of human skeletal muscle protein synthesis by essential amino acids. J. Nutr. 2011, 141, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Baar, E.L.; Carbajal, K.A.; Ong, I.M.; Lamming, D.W. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell 2016, 15, 155–166. [Google Scholar] [CrossRef]
- Goldstein, I.; Hager, G.L. Transcriptional and Chromatin Regulation during Fasting—The Genomic Era. Trends Endocrinol. Metab. 2015, 26, 699–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, A.L.; Crane, J.D.; Lally, J.S.; Ford, R.J.; Kang, S.J.; Rebalka, I.A.; Green, A.E.; Kemp, B.E.; Hawke, T.J.; Schertzer, J.D.; et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015, 21, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.D.; Ji, X.; et al. mTORC1 underlies age-related muscle fiber damage and loss by inducing oxidative stress and catabolism. Aging Cell 2019, 18, e12943. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Landum, R.W.; Cheng, M.S.; Wu, J.F.; Floyd, R.A. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha-phenylnitrone. Proc. Natl. Acad. Sci. USA 1991, 88, 3633–3636. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- De Graff, A.M.; Hazoglou, M.J.; Dill, K.A. Highly Charged Proteins: The Achilles’ Heel of Aging Proteomes. Structure 2016, 24, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Starke-Reed, P.E.; Oliver, C.N. Protein oxidation and proteolysis during aging and oxidative stress. Arch Biochem. Biophys. 1989, 275, 559–567. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K. Inclusion-body myositis: Muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer’s and Parkinson’s disease brains. Acta Neuropathol. 2008, 116, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [Green Version]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Altun, M.; Besche, H.C.; Overkleeft, H.S.; Piccirillo, R.; Edelmann, M.J.; Kessler, B.M.; Goldberg, A.L.; Ulfhake, B. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J. Biol. Chem. 2010, 285, 39597–39608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, V.; Baracos, V.; Sarraf, P.; Goldberg, A.L. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 12602–12607. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.Z.; Drouin, G.; Desrosiers, J.; Trensz, F.; Grenier, G. A novel hindlimb immobilization procedure for studying skeletal muscle atrophy and recovery in mouse. J. Appl. Physiol. (1985) 2009, 106, 2049–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, T.; Furochi, H.; Mameoka, M.; Hirasaka, K.; Onishi, Y.; Suzue, N.; Oarada, M.; Akamatsu, M.; Akima, H.; Fukunaga, T.; et al. Ubiquitin ligase gene expression in healthy volunteers with 20-day bedrest. Muscle Nerve 2006, 34, 463–469. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007, 21, 140–155. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Hunt, L.C.; Schadeberg, B.; Stover, J.; Haugen, B.; Pagala, V.; Wang, Y.D.; Puglise, J.; Barton, E.R.; Peng, J.; Demontis, F. Antagonistic control of myofiber size and muscle protein quality control by the ubiquitin ligase UBR4 during aging. Nat. Commun. 2021, 12, 1418. [Google Scholar] [CrossRef]
- Kitajima, Y.; Suzuki, N.; Yoshioka, K.; Izumi, R.; Tateyama, M.; Tashiro, Y.; Takahashi, R.; Aoki, M.; Ono, Y. Inducible Rpt3, a Proteasome Component, Knockout in Adult Skeletal Muscle Results in Muscle Atrophy. Front. Cell Dev. Biol. 2020, 8, 859. [Google Scholar] [CrossRef]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M.; et al. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Bargiela, A.; Cerro-Herreros, E.; Fernandez-Costa, J.M.; Vilchez, J.J.; Llamusi, B.; Artero, R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis. Model. Mech. 2015, 8, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef]
- Zhang, F.; Romano, P.R.; Nagamura-Inoue, T.; Tian, B.; Dever, T.E.; Mathews, M.B.; Ozato, K.; Hinnebusch, A.G. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 2001, 276, 24946–24958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanduri, S.; Rahman, F.; Williams, B.R.; Qin, J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000, 19, 5567–5574. [Google Scholar] [CrossRef] [Green Version]
- Zhan, K.; Vattem, K.M.; Bauer, B.N.; Dever, T.E.; Chen, J.J.; Wek, R.C. Phosphorylation of eukaryotic initiation factor 2 by heme-regulated inhibitor kinase-related protein kinases in Schizosaccharomyces pombe is important for fesistance to environmental stresses. Mol. Cell Biol. 2002, 22, 7134–7146. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol. Cell Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Qiu, H.; Garcia-Barrio, M.; Anderson, J.; Hinnebusch, A.G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 2000, 6, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654.e612. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Waddell, D.S.; Baehr, L.M.; van den Brandt, J.; Johnsen, S.A.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E785–E797. [Google Scholar] [CrossRef]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.T.; Bhardwaj, G.; Penniman, C.M.; Krumpoch, M.T.; Suarez Beltran, P.A.; Klaus, K.; Poro, K.; Li, M.; Pan, H.; Dreyfuss, J.M.; et al. FoxO Transcription Factors Are Critical Regulators of Diabetes-Related Muscle Atrophy. Diabetes 2019, 68, 556–570. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, M.J.; Foley, K.P.; D’Souza, D.M.; Li, Y.E.; Lau, T.C.; Hawke, T.J.; Schertzer, J.D. The NLRP3 inflammasome contributes to sarcopenia and lower muscle glycolytic potential in old mice. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E222–E232. [Google Scholar] [CrossRef] [Green Version]
- Dirks, A.; Leeuwenburgh, C. Apoptosis in skeletal muscle with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R519–R527. [Google Scholar] [CrossRef] [Green Version]
- Price, S.R.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; England, B.K.; Ding, X.; Phillips, L.S.; Mitch, W.E. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Investig. 1996, 98, 1703–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Plant, P.J.; Bain, J.R.; Correa, J.E.; Woo, M.; Batt, J. Absence of caspase-3 protects against denervation-induced skeletal muscle atrophy. J. Appl. Physiol. (1985) 2009, 107, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belizario, J.E.; Lorite, M.J.; Tisdale, M.J. Cleavage of caspases-1, -3, -6, -8 and -9 substrates by proteases in skeletal muscles from mice undergoing cancer cachexia. Br. J. Cancer 2001, 84, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C. Sporadic inclusion body myositis--diagnosis, pathogenesis and therapeutic strategies. Nat. Clin. Pract. Neurol. 2006, 2, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, W.K.; Askanas, V. Inclusion-body myositis: Clinical, diagnostic, and pathologic aspects. Neurology 2006, 66, S20–S29. [Google Scholar] [CrossRef]
- Salajegheh, M.; Pinkus, J.L.; Taylor, J.P.; Amato, A.A.; Nazareno, R.; Baloh, R.H.; Greenberg, S.A. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 2009, 40, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Weihl, C.C.; Pestronk, A. Sporadic inclusion body myositis: Possible pathogenesis inferred from biomarkers. Curr. Opin. Neurol. 2010, 23, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.S.; Weihl, C.C. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: A disorder of autophagy. Hum. Mol. Genet 2010, 19, R38–R45. [Google Scholar] [CrossRef] [PubMed]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef]
- Fratta, P.; Engel, W.K.; McFerrin, J.; Davies, K.J.; Lin, S.W.; Askanas, V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am. J. Pathol. 2005, 167, 517–526. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Custer, S.K.; Neumann, M.; Lu, H.; Wright, A.C.; Taylor, J.P. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet 2010, 19, 1741–1755. [Google Scholar] [CrossRef]
- Ahmed, M.; Machado, P.M.; Miller, A.; Spicer, C.; Herbelin, L.; He, J.; Noel, J.; Wang, Y.; McVey, A.L.; Pasnoor, M.; et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci. Transl. Med. 2016, 8, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, W. RNAi: The nuts and bolts of the RISC machine. Cell 2005, 122, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, J.; Lasko, P. Translational control in cellular and developmental processes. Nat. Rev. Genet. 2012, 13, 383–394. [Google Scholar] [CrossRef]
- Rajkowitsch, L.; Chen, D.; Stampfl, S.; Semrad, K.; Waldsich, C.; Mayer, O.; Jantsch, M.F.; Konrat, R.; Blasi, U.; Schroeder, R. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007, 4, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Rabinovitch, P.S. The Aging Heart. Cold Spring Harb. Perspect. Med. 2015, 5, a025148. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Nicotera, P. MicroRNAs in age-related diseases. EMBO Mol. Med. 2013, 5, 180–190. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, C.; Komici, K.; Borghetti, G.; Femminella, G.D.; Bencivenga, L.; Cannavo, A.; Corbi, G.; Ferrara, N.; Houser, S.R.; Koch, W.J.; et al. microRNA in Cardiovascular Aging and Age-Related Cardiovascular Diseases. Front. Med. 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, P.; Montano, M.; Puca, A.; Solovieff, N.; Kojima, T.; Wang, M.C.; Melista, E.; Meltzer, M.; Fischer, S.E.; Andersen, S.; et al. RNA editing genes associated with extreme old age in humans and with lifespan in C. elegans. PLoS ONE 2009, 4, e8210. [Google Scholar] [CrossRef]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.; Kawahara, Y.; Tamburro, K.M.; Nishikura, K. A-to-I RNA editing and human disease. RNA Biol. 2006, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Muller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Piec, I.; Listrat, A.; Alliot, J.; Chambon, C.; Taylor, R.G.; Bechet, D. Differential proteome analysis of aging in rat skeletal muscle. FASEB J. 2005, 19, 1143–1145. [Google Scholar] [CrossRef]
- Gan, Z.; Zhao, L.; Yang, L.; Huang, P.; Zhao, F.; Li, W.; Liu, Y. RNA editing by ADAR2 is metabolically regulated in pancreatic islets and beta-cells. J. Biol. Chem. 2006, 281, 33386–33394. [Google Scholar] [CrossRef] [Green Version]
- Bavelloni, A.; Focaccia, E.; Piazzi, M.; Raffini, M.; Cesarini, V.; Tomaselli, S.; Orsini, A.; Ratti, S.; Faenza, I.; Cocco, L.; et al. AKT-dependent phosphorylation of the adenosine deaminases ADAR-1 and -2 inhibits deaminase activity. FASEB J. 2019, 33, 9044–9061. [Google Scholar] [CrossRef]
- Picardi, E.; D’Erchia, A.M.; Lo Giudice, C.; Pesole, G. REDIportal: A comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017, 45, D750–D757. [Google Scholar] [CrossRef] [Green Version]
- Ramaswami, G.; Li, J.B. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014, 42, D109–D113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, M.; Futreal, P.A.; Wooster, R.; Stratton, M.R. A survey of RNA editing in human brain. Genome Res. 2004, 14, 2379–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschenes, M.; Chabot, B. The emerging role of alternative splicing in senescence and aging. Aging Cell 2017, 16, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W.B. Alternative splicing in aging and longevity. Hum. Genet. 2020, 139, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.P.; Pilling, L.C.; Emond, F.; Flurkey, K.; Harrison, D.E.; Yuan, R.; Peters, L.L.; Kuchel, G.A.; Ferrucci, L.; Melzer, D.; et al. Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell 2016, 15, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Zahn, J.M.; Sonu, R.; Vogel, H.; Crane, E.; Mazan-Mamczarz, K.; Rabkin, R.; Davis, R.W.; Becker, K.G.; Owen, A.B.; Kim, S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006, 2, e115. [Google Scholar] [CrossRef]
- Rodriguez, S.A.; Grochova, D.; McKenna, T.; Borate, B.; Trivedi, N.S.; Erdos, M.R.; Eriksson, M. Global genome splicing analysis reveals an increased number of alternatively spliced genes with aging. Aging Cell 2016, 15, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Wu, D.; Zhang, H.; Das, A.; Basu, M.; Malin, J.; Cao, K.; Hannenhalli, S. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Sci. Rep. 2018, 8, 10929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollervey, J.R.; Wang, Z.; Hortobagyi, T.; Witten, J.T.; Zarnack, K.; Kayikci, M.; Clark, T.A.; Schweitzer, A.C.; Rot, G.; Curk, T.; et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 2011, 21, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Bill, B.R.; Lowe, J.K.; Dybuncio, C.T.; Fogel, B.L. Orchestration of neurodevelopmental programs by RBFOX1: Implications for autism spectrum disorder. Int. Rev. Neurobiol. 2013, 113, 251–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, L.K.; Maltman, N.; Mosconi, M.W.; Macmillan, C.; Schmitt, L.; Moore, K.; Francis, S.M.; Jacob, S.; Sweeney, J.A.; Cook, E.H. Rare inherited A2BP1 deletion in a proband with autism and developmental hemiparesis. Am. J. Med. Genet. A 2012, 158A, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Pedrotti, S.; Giudice, J.; Dagnino-Acosta, A.; Knoblauch, M.; Singh, R.K.; Hanna, A.; Mo, Q.; Hicks, J.; Hamilton, S.; Cooper, T.A. The RNA-binding protein Rbfox1 regulates splicing required for skeletal muscle structure and function. Hum. Mol. Genet. 2015, 24, 2360–2374. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Kolonin, A.M.; Fiorotto, M.L.; Cooper, T.A. Rbfox-Splicing Factors Maintain Skeletal Muscle Mass by Regulating Calpain3 and Proteostasis. Cell Rep. 2018, 24, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Runfola, V.; Sebastian, S.; Dilworth, F.J.; Gabellini, D. Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J. Cell Sci. 2015, 128, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat. Commun. 2018, 9, 2010. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Iida, K.; Tsubota, T.; Hosokawa, M.; Denawa, M.; Brown, J.B.; Ninomiya, K.; Ito, M.; Kimura, H.; Abe, T.; et al. Loss of Sfpq Causes Long-Gene Transcriptopathy in the Brain. Cell Rep. 2018, 23, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Takeuchi, A.; Tanihata, J.; Iida, K.; Takeda, S.; Hagiwara, M. Loss of RNA-Binding Protein Sfpq Causes Long-Gene Transcriptopathy in Skeletal Muscle and Severe Muscle Mass Reduction with Metabolic Myopathy. iScience 2019, 13, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef]
- Kwong, L.K.; Sohal, R.S. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch Biochem. Biophys. 2000, 373, 16–22. [Google Scholar] [CrossRef]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: A review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef] [Green Version]
- Leduc, M.S.; Chao, H.T.; Qu, C.; Walkiewicz, M.; Xiao, R.; Magoulas, P.; Pan, S.; Beuten, J.; He, W.; Bernstein, J.A.; et al. Clinical and molecular characterization of de novo loss of function variants in HNRNPU. Am. J. Med. Genet. A 2017, 173, 2680–2689. [Google Scholar] [CrossRef]
- Durkin, A.; Albaba, S.; Fry, A.E.; Morton, J.E.; Douglas, A.; Beleza, A.; Williams, D.; Volker-Touw, C.M.L.; Lynch, S.A.; Canham, N.; et al. Clinical findings of 21 previously unreported probands with HNRNPU-related syndrome and comprehensive literature review. Am. J. Med. Genet. A 2020, 182, 1637–1654. [Google Scholar] [CrossRef] [Green Version]
- Depienne, C.; Nava, C.; Keren, B.; Heide, S.; Rastetter, A.; Passemard, S.; Chantot-Bastaraud, S.; Moutard, M.L.; Agrawal, P.B.; VanNoy, G.; et al. Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum. Genet. 2017, 136, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Wee, C.D.; Havens, M.A.; Jodelka, F.M.; Hastings, M.L. Targeting SR proteins improves SMN expression in spinal muscular atrophy cells. PLoS ONE 2014, 9, e115205. [Google Scholar] [CrossRef]
- Xiao, R.; Tang, P.; Yang, B.; Huang, J.; Zhou, Y.; Shao, C.; Li, H.; Sun, H.; Zhang, Y.; Fu, X.D. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell 2012, 45, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Lv, P.; Huo, X.; Wu, J.; Wang, Q.; Cheng, L.; Liu, Y.; Tang, Q.Q.; Zhang, L.; Zhang, F.; et al. The nuclear matrix protein HNRNPU maintains 3D genome architecture globally in mouse hepatocytes. Genome Res. 2018, 28, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, Y.; Brockdorff, N.; Kawano, S.; Tsutui, K.; Tsutui, K.; Nakagawa, S. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell 2010, 19, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.L.; Banerjee, S.; Hegde, P.M.; Bellot, L.J.; Hazra, T.K.; Boldogh, I.; Mitra, S. Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 2012, 287, 34202–34211. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Beetz, N.; O’Keeffe, S.; Tapia, J.C.; Macpherson, L.; Chen, W.V.; Bassel-Duby, R.; Olson, E.N.; Maniatis, T. hnRNP U protein is required for normal pre-mRNA splicing and postnatal heart development and function. Proc. Natl. Acad. Sci. USA 2015, 112, E3020–E3029. [Google Scholar] [CrossRef] [Green Version]
- Seino, S.; Bell, G.I. Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 1989, 159, 312–316. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413. [Google Scholar] [CrossRef]
- Sen, S.; Talukdar, I.; Webster, N.J. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell Biol. 2009, 29, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, I.; Sen, S.; Urbano, R.; Thompson, J.; Yates, J.R., 3rd; Webster, N.J. hnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS ONE 2011, 6, e27869. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, G.V.; Cooper, T.A. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 2014, 42, 1893–1903. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Dansithong, W.; Kim, D.; Rossi, J.; Webster, N.J.; Comai, L.; Reddy, S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006, 25, 4271–4283. [Google Scholar] [CrossRef] [Green Version]
- Jansen, G.; Mahadevan, M.; Amemiya, C.; Wormskamp, N.; Segers, B.; Hendriks, W.; O’Hoy, K.; Baird, S.; Sabourin, L.; Lennon, G.; et al. Characterization of the myotonic dystrophy region predicts multiple protein isoform-encoding mRNAs. Nat. Genet. 1992, 1, 261–266. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Cooper, T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A.; Dalton, J.C.; Moseley, M.L.; Ranum, L.P.; Day, J.W. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am. J. Hum. Genet. 2004, 74, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, M.; Masciullo, M.; Bonvissuto, D.; Bianchi, M.L.; Michetti, F.; Silvestri, G. Alternative splicing of human insulin receptor gene (INSR) in type I and type II skeletal muscle fibers of patients with myotonic dystrophy type 1 and type 2. Mol. Cell Biochem. 2013, 380, 259–265. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Mosthaf, L.; Eriksson, J.; Haring, H.U.; Groop, L.; Widen, E.; Ullrich, A. Insulin receptor isotype expression correlates with risk of non-insulin-dependent diabetes. Proc. Natl. Acad. Sci. USA 1993, 90, 2633–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benecke, H.; Flier, J.S.; Moller, D.E. Alternatively spliced variants of the insulin receptor protein. Expression in normal and diabetic human tissues. J. Clin. Investig. 1992, 89, 2066–2070. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.; Bjorbaek, C.; Vestergaard, H.; Gronskov, K.; Bak, J.F.; Pedersen, O. Expression of insulin receptor spliced variants and their functional correlates in muscle from patients with non-insulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1993, 77, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Mosthaf, L.; Vogt, B.; Haring, H.U.; Ullrich, A. Altered expression of insulin receptor types A and B in the skeletal muscle of non-insulin-dependent diabetes mellitus patients. Proc. Natl. Acad. Sci. USA 1991, 88, 4728–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passacantilli, I.; Frisone, P.; De Paola, E.; Fidaleo, M.; Paronetto, M.P. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 2017, 45, 12270–12284. [Google Scholar] [CrossRef]
- Chen, W.Y.; Lin, C.L.; Chuang, J.H.; Chiu, F.Y.; Sun, Y.Y.; Liang, M.C.; Lin, Y. Heterogeneous nuclear ribonucleoprotein M associates with mTORC2 and regulates muscle differentiation. Sci. Rep. 2017, 7, 41159. [Google Scholar] [CrossRef]
- Ghosh, D.; Srivastava, G.P.; Xu, D.; Schulz, L.C.; Roberts, R.M. A link between SIN1 (MAPKAP1) and poly(rC) binding protein 2 (PCBP2) in counteracting environmental stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11673–11678. [Google Scholar] [CrossRef] [Green Version]
- Karni, R.; Hippo, Y.; Lowe, S.W.; Krainer, A.R. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc. Natl. Acad. Sci. USA 2008, 105, 15323–15327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michlewski, G.; Sanford, J.R.; Caceres, J.F. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol. Cell 2008, 30, 179–189. [Google Scholar] [CrossRef]
- United Nations. World Population Ageing 2019: Highlights; United Nations: New York, NY, USA, 2019. [Google Scholar]
- Janssen, I.; Baumgartner, R.N.; Ross, R.; Rosenberg, I.H.; Roubenoff, R. Skeletal muscle cutpoints associated with elevated physical disability risk in older men and women. Am. J. Epidemiol. 2004, 159, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Paddon-Jones, D.; Short, K.R.; Campbell, W.W.; Volpi, E.; Wolfe, R.R. Role of dietary protein in the sarcopenia of aging. Am. J. Clin. Nutr. 2008, 87, 1562S–1566S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Signaling pathways downstream of insulin/IGF-1 receptor activation and crosstalk with catabolic signaling pathways. Signaling pathways observed in multiple cells lines and tissues in vivo (black lines). Signaling pathways observed in cancer cells resistant to PI3K inhibitors (grey lines).
Figure 1.
Signaling pathways downstream of insulin/IGF-1 receptor activation and crosstalk with catabolic signaling pathways. Signaling pathways observed in multiple cells lines and tissues in vivo (black lines). Signaling pathways observed in cancer cells resistant to PI3K inhibitors (grey lines).

Figure 2.
Nutrient sensing pathways modulate protein synthesis. Cells respond to cellular stress signals to both inhibit protein synthesis and increase the expression of genes to alleviate cellular stress.
Figure 2.
Nutrient sensing pathways modulate protein synthesis. Cells respond to cellular stress signals to both inhibit protein synthesis and increase the expression of genes to alleviate cellular stress.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, E.-J.; Neppl, R.L. Influence of Age on Skeletal Muscle Hypertrophy and Atrophy Signaling: Established Paradigms and Unexpected Links. Genes 2021, 12, 688. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050688
AMA Style
Lee E-J, Neppl RL. Influence of Age on Skeletal Muscle Hypertrophy and Atrophy Signaling: Established Paradigms and Unexpected Links. Genes. 2021; 12(5):688. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050688
Chicago/Turabian StyleLee, Eun-Joo, and Ronald L. Neppl. 2021. "Influence of Age on Skeletal Muscle Hypertrophy and Atrophy Signaling: Established Paradigms and Unexpected Links" Genes 12, no. 5: 688. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050688
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.