1. Introduction
X-linked adrenoleukodystrophy (X-ALD, OMIM #300100) is the most common peroxisomal disorder. The disease is caused by defects in the gene for the adenosine triphosphate (ATP)-binding cassette protein, subfamily D (
ABCD1) [
1,
2,
3,
4]. This gene encodes the peroxisomal transporter of very-long-chain fatty acids (VLCFAs) [
5,
6]. The defective function of
ABCD1 protein prevents β-oxidation of VLCFAs, leading to accumulation in tissues and plasma, which represents the disease hallmark [
7].
X-ALD is clinically characterized by two main phenotypes: adrenomyeloneuropathy (AMN) and the cerebral demyelinating form of X-ALD (cerebral ALD) [
1,
8,
9].
Cerebral ALD presents usually with a rapidly progressive inflammatory demyelination of the cerebral white matter, which produces severe cognitive and motor disability in childhood (childhood cerebral ALD; CCALD) and adolescence (adolescent cerebral ALD) [
9].
The pathology of AMN is distinct from cerebral ALD and it is mostly characterized by a non-inflammatory distal axonopathy, which mostly affects the long descending pathways in the white matter of the spinal cord, with an emphasis on the corticospinal pathway, which leads to progressive spastic para-paresis [
1,
8,
9].
As in many X-linked diseases, it was assumed that female carriers remain asymptomatic until it was reported that also heterozygous females eventually, as time goes by, suffer from AMN symptoms [
10,
11,
12,
13]. Moreover, these female patients, even in the early asymptomatic stage, already carry some abnormalities, which can be detected by instrumental neurophysiology [
5]. Sometimes, at this stage also adrenal failure and cerebral adrenoleukodystrophy may appear [
5,
14,
15,
16,
17].
Recent clinical evaluations in prospective cross-sectional cohort studies have detected neurological impairment in females, reporting the evolution of phenotypes with aging [
5,
18]. These studies demonstrated that carriers develop an adrenomyeloneuropathy-like phenotype with a strong association between symptomatic status and age [
5]. Symptoms include myelopathy (63%) and/or peripheral neuropathy (57%) [
5].
Generally, the clinical phenotype of carrier women is milder than in men, with a typical onset after the fourth decade. However, some exceptions have been reported and sometimes a severe phenotype can be recognized in carrier women with early-onset [
19].
Based on mutational analysis of the
ABCD1 gene in X-ALD patients, about 600 different mutations have been described so far [
20] (
http://www.x-ald.nl, accessed on 10 October 2020).
According to the known intra-familial variability, it was supposed that other (epi)genetic factors contribute to the phenotype of patients with
ABCD1 mutations [
21].
Therefore, although large deletions, nonsense or frameshift mutations that result in the complete absence of a functional ALDP have been found in patients covering the full spectrum of X-ALD phenotypes, no strong genotype-phenotype correlation has been described. In clinical practice, the absence of a strong genetic relationship with the phenotype makes difficult the neurological characterization, leading often to misdiagnosis.
In this paper, we report on a large family in which affected males died during the first decade, while affected females developed symptoms within the fourth decade with spastic or ataxic-spastic gait, tetra-hyperreflexia, lower limbs hyposthenia, and sensory complaints.
2. Materials and Methods
2.1. Ethics
A written informed consent for genetic analyses was obtained from the patients or patients’ parents. The research work was carried out following ethical principles and the Italian legislation. The study was approved by IRCCS Neuromed ethical committees. The study is registered on ClinicalTrials.gov (NCT03084224).
2.2. Demographic and Clinical Features
This paper describes a five-generation family in which two main phenotypes of X-ALD are segregated (
Figure 1). The affected male died in the first decade (III:4; III:8; III:13; and III:14), and all affected females present with gait disturbance and different neurological features (
Figure 1,
Table 1). The genetic evaluation included nine people, including eight females and one male. Of the eight females, five are affected (III:2; III:6; III:10; IV:2; and IV:4), age at onset ranging between 34 and 42 years (average 38 years); and 3 are healthy (IV:8; V:1; and V:2), age at evaluation ranging between 26 and 39 years (average 31 years). The healthy male IV:5 is 35 years old.
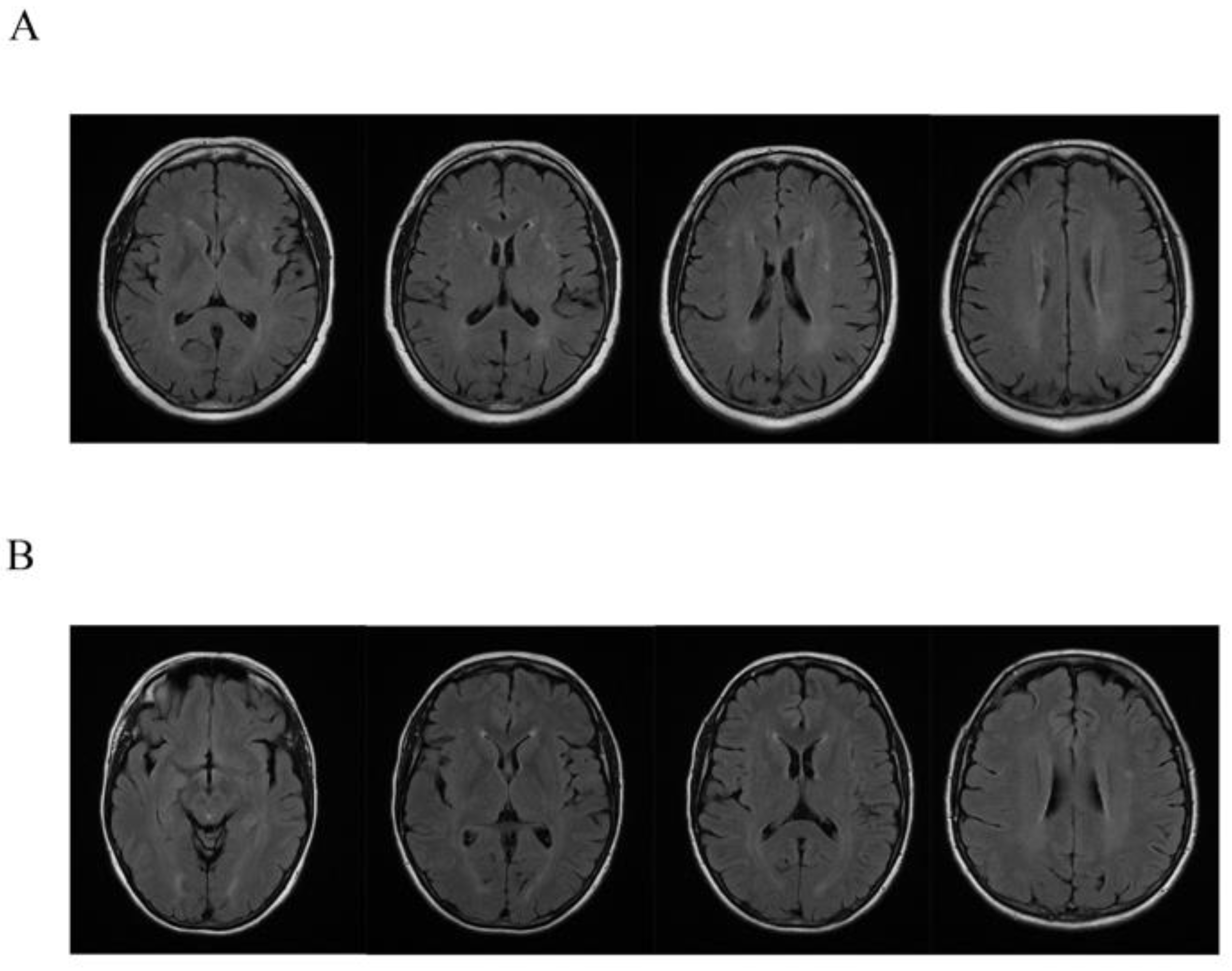
Clinical, neurological examination and brain MRI scan were performed in the affected female (
Table 1,
Figure 2). A complete neuropsychological evaluation was performed in patients IV:2 and IV:4 and a psychological interview in patient III:3.
2.3. Genetic Analysis
Genomic DNA was isolated from peripheral blood leukocytes according to standard procedures (QIAamp DNA Blood Mini Kit–QIAGEN). Clinical exome sequencing considering about 5000 human genes was performed using the Clinical Exome Solution kit (Sophia Genetics, SA, Boston, MA, USA), following the manufacturer’s instructions. The resulting libraries were processed for paired-end sequencing on the MiSeq platform Illumina (San Diego, CA, USA). Sophia DDM® platform (Sophia Genetics, SA) was used for automated annotation, characterization, and selection of potentially pathogenic variants. Direct evaluation of the data sequence was performed by the Integrative Genomics Viewer v.2.3.
A second analysis by using GenomeUp platfarm was performed (
https://platform.genomeup.com/, accessed on 10 October 2020) using the Best Practices workflows of GATK v4.1 for germline variant calling.
Potentially pathogenic variants were interpreted according to ACMG criteria [
22]. ACMG classification was compared with automatic classification performed by Varsome genome interpreter (
https://varsome.com/, accessed on 10 October 2020).
Variant NM_000033:c.[1661G>A]; NP_000024.2:p.(Arg554His) rs201568579 (ClinVar # VCV000166625) in ABCD1 gene was selected as potentially responsible for the clinical phenotype (class 5, PP5-PM1-PM2-PM5-PP2-PP3).
Mutation re-sequencing and segregation analysis were performed by Sanger sequencing ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). To perform direct re-sequencing of the pathogenic variant in ABCD1, sequence-specific primers were designed (FW–5′ GAGTATCTTGGGGGAGGCAG 3′; RW–5′ ATCTGTGTGGTGTTGGTCCTC 3′) to avoid amplification of homolog sequences of the ABCD1 pseudogenes at chromosomes 2p11, 10p11, 16p11, and 22q11 (92–96% of similarities with exons 7–10 of ABCD1 gene).
2.4. Biochemical Analysis
Blood samples were collected to measure plasma levels of the VLCFA C22:0, C24:0, and C26:0. VLCFA values of C26:0, of C26:0/C22:0, and C24:0/C22:0 were evaluated. Details of all these methods were described in Habekost et al., 2014 [
18].
2.5. Literature Review of p.(Arg554His) Variant
A systematic review of the literature was conducted to identify studies reporting male and female patients with p.(Arg554His) mutation. The literature search identified 14 publications describing 23 patients. The gender is reported for 13 male patients. The phenotype reported in association with this variant includes ALD, CCALD, AMN, and asymptomatic carrier (
Table 2).
2.6. Statistic and Data Analysis
An unpaired t-test was conducted comparing reference values with plasma values of C26:0, C26:0/C22:0, and C26:0/C24:0 ratio in female carriers vs. healthy male.
3. Results
Probands III:2 were referred to the Neurology Unit of Neuromed IRCCS (Italy), for gait difficulties, imbalance, and lower limb stiffness. Familial anamnesis revealed that she is part of a large family in which affected males died with the first decade, while affected females showed progressive spastic or ataxic-spastic gait, tetra-hyperreflexia, lower limbs hyposthenia, and sensory complaints.
Sequencing analysis performed on III:2 identified a pathogenic variant in ABCD1 gene (NM_000033:c.[1661G>A]; NP_000024.2:p.(Arg554His); rs201568579). This variant is responsible for X-linked adrenoleukodystrophy (OMIM #300100), and it is has been described in male patients affected by X-ALD.
To fully understand the role of this variant in the clinical phenotype of the numerous affected patients, genetic analyses were performed in four more symptomatic females (III:6; III:10; IV:2; IV:4), confirming the segregation of the identified mutation with the pathological phenotype. To further confirm the segregation, a sequencing analysis on a healthy son (IV:5) of an affected woman (III:6) was performed. The analysis revealed the absence of mutation, confirming the inheritance of the maternal wild-type allele.
VLFA dosage was conducted in patients III:2; III:6; III:10; IV:2; IV:4; and IV:8. Plasma C26:0 levels, C24:0/C22:0, and C26:0/C22:0 ratios were increased in all the female samples (
Table 1, C26:0 levels
p = 0.0078; C26:0/C22:0
p = 0.0127).
The female symptomatic carriers (III:2; III:6; III:10; IV:2 and IV:4) were clinically characterized (
Table 1).
The age of symptoms ranged between 34 and 42 years, with an average of 38 years. All the women showed first progressive lower limb weakness with gait difficulties, imbalance, sometimes lower limb stiffness and, in one case, cramps or paraesthesia. Sphincter disturbances were present in varying degrees. Neurological examination showed spastic or ataxic-spastic gait, tetra-hyperreflexia, Babinski sign, lower limbs hyposthenia, sensory complaints (lower limb hypopallesthesia or pain), and in some cases diplopia or light upper limb dysmetria. No patients had hyperpigmentation or another adrenal symptom of adrenal failure. All of them showed multiple discopathy of the spinal cord without myelopathy.
Complete cognitive profile was assessed in patients IV:2 and IV:4. The first showed a slight impairment in praxic-constructive and logical-deductive skills, severe depression, and anxiety with conversion aspects. Patient IV:4 had a normal cognitive profile, moderate depression, and anxiety. The psychological interview of patient III:3 also showed the presence of depression and anxiety.
Neurophysiological analysis was conducted in all affected females. There was no relief of peripheral neuropathy at nerve conduction studies (ENG) and electromyography (EMG). Motor evoked potentials (MEPs) of the lower limbs always showed bilateral, an asymmetric increase of central motor conduction time at lower limbs. Likewise, somatosensory evoked potentials (SEP) showed bilateral, an asymmetric increase of central conduction time at lower limbs. Brainstem auditory evoked potentials (BAEPs) were available for patients III:2 and IV:2. Patient III:2 showed a normal response. Patient IV:2 had bilateral and asymmetric dysfunctions of the auditory pathways. The visual evoked response was normal in all the analyzed cases (III:10; IV:2; IV:4).
Brain 1,5 tesla MRI in all affected female carriers showed some and small diffuse T2 hyperintensity localized in the context of the white matter of both cerebral hemispheres. Concomitant nuanced hyperintensity in the same sequences of the periventricular white matter were detected (
Figure 2).
4. Discussion
As in many X-linked diseases, in adrenoleukodystrophy, it is expected that female carriers remain asymptomatic. Recent papers reported that most ABCD1 female carriers indeed show at first instrumental abnormalities and, later on, frank neurological symptoms. Thus, what was once considered just a simple carrier, now develops a clinical phenotype reminiscent of a frank adrenomyeloneuropathy including cognitive impairment and motor disability. The natural disease progression of ABCD1 heterozygous female carriers is strongly age-related since roughly 80% of the female carriers carry some symptom beyond the age of 60. Contrariwise to AMN where adult males show complete penetrance, female carriers show a reduced penetrance with a cut-off age at about 40 years critical for phenotypic conversion.
Data concerning female carriers have been produced by cross-sectional cohort studies or longitudinal data retrospectively reviewed. These data provided a detailed description of clinical and neurological phenotype in females but failed to focus on the genotype-phenotype correlation.
Conversely, in this paper, by profiting off a study that was conducted in a large family affected by X-ALD, such a correlation was addressed. In this family, all affected males died within the first decade. Female carriers are heterozygote for the mutation p.(Arg554His) in the
ABCD1 gene. This mutation is pathogenic considering the AMG classification and the disease-specific mutation database available at
www.x-ald.nl, accessed on 10 October 2020.
This variant, mainly reported in cross-sectional analysis considering X-ALD male patients, is not described in female carriers, and genotype/phenotype has never been evaluated. Our systematic search of the literature conducted to identify carriers of p.(Arg554His) mutation shows that male patient carriers of this variant suffer from ALD, CCALD, and AMN (
Table 2).
This is in line with other papers reporting the lack of genotype/phenotype in X-ALD correlation in males. The common frameshift mutation (p.Gln472fsX83) leading to a truncated ALDP identified in 81 patients, and the p.Pro484Arg identified in a family with six male patients, presented with a wide clinical variability of X-ALD [
36,
37].
Another paper by Margoni et al., 2017 [
38] highlighted the wide range of phenotypic expressions of ALD, reporting a novel heterozygous mutation IVS4+2T>A (c.1393+2T>A) in a family with six members (two females) carrying different phenotypes of the entire clinical and radiological spectrum of X-ALD.
Females heterozygous for X-ALD can develop a wide range of neurologic abnormalities, most of them consisting of an AMN-like phenotype, and neurological impairment may finally affect most (if not all) of them and progress with age in severity, independently from the
ABCD1 variant [
39].
Therefore, studies concerning large families of X-ALD considering genotype/phenotype in females are required.
In this report, we analyzed a large family affected by X-ALD, with five female carriers of p.(Arg554His) mutations and developing full penetrance within 42 years. All the women showed at first progressive spastic or ataxic-spastic gait, tetra-hyperreflexia, lower limbs hyposthenia, and sensory complaints. Evoked motor and somatosensory potentials were affected abnormally in all the female carriers, confirming the involvement of the CNS at the level of the spinal cord.
The neuropsychological assessment showed deficits in praxic-constructive and logical-deductive skills in one patient while a depression/anxiety spectrum was evident in all tested patients. Cognitive impairment and/or psychiatric symptoms in our female population suggest the presence of brain involvement in AMN-like phenotype.
While the neuroimaging findings of cerebral ALD and AMN have been well described in men, the same findings in women are less investigated [
2]. We found a non-specific 1.5 T brain MRI pattern in all the symptomatic female carriers characterized by more or less diffuse T2 hyperintense lesions in white matter associated with nuanced periventricular hyperintensity. Although these represent non-specific and nuanced changes, the carriers did not have the common cardiovascular risk factors, so these lesions cannot be explained otherwise and must be taken into account.
This framework suggests giving more attention to routine brain MRI in suspected X-ALD carriers, also in AMN-like phenotype, to detect the slight and non-specific pattern and to better define neuroimaging findings in female carriers.
5. Conclusions
In this study, we describe a large family that allowed us to study the role of an ABCD1 mutation in five female carriers that developed a full penetrance within 42 years. These allowed to strengthen the relevance of clinical symptoms in the female, which leads to a better understanding of the role of the genetic background.
Although a genotype-phenotype correlation in male patients has not been established, the data suggest the relevance of obtaining information concerning this association in female carriers.
Moreover, these data indicate the relevance to include ABCD1 genes in genetic panels for gait disturbance in women.
Author Contributions
D.C., C.F., F.B. (Fabio Buttari), S.Z., C.D. and performed the recruitment and clinical evaluations of patients. R.F., R.C., M.A.C., and F.B. (Francesca Biagioni) performed the genetic analyses. D.C., M.F., S.A., E.G., F.F., M.S. and S.G. supervised the work. R.C. and S.G. wrote the first draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Italian Ministry of Health (Current Research 2019–2023: Identification of New Variants and/or New Genes Responsible for Ataxia and Spastic Paraplegia), (5XMille–2018).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of IRCCS Neuromed. The study is registered on ClinicalTrials.gov (NCT03084224).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Acknowledgments
The authors are grateful to the patients and their relatives participating in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moser, H.W.; Mahmood, A.; Raymond, G.V. X-linked adrenoleukodystrophy. Nat. Clin. Pract. Neurol. 2007, 3, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Jangouk, P.; Zackowski, K.M.; Naidu, S.; Raymond, G.V. Adrenoleukodystrophy in female heterozygotes: Underrecognized and undertreated. Mol. Genet. Metab. 2012, 105, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet. J. Rare Dis. 2012, 13, 51. [Google Scholar] [CrossRef]
- Horn, M.A.; Retterstøl, L.; Abdelnoor, M.; Skjeldal, O.H.; Tallaksen, C.M. Adrenoleukodystrophy in Norway: High rate of de novo mutations and age-dependent penetrance. Pediatr. Neurol. 2013, 48, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelen, M.; Barbier, M.; Dijkstra, I.M.; Schür, R.; de Bie, R.M.; Verhamme, C.; Dijkgraaf, M.G.; Aubourg, P.A.; Wanders, R.J.; van Geel, B.M.; et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain 2014, 137, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habekost, C.T.; Pereira, F.S.; Vargas, C.R.; Coelho, D.M.; Torrez, V.; Oses, J.P.; Portela, L.V.; Schestatsky, P.; Felix, V.T.; Matte, U.; et al. Progression rate of myelopathy in X-linked adrenoleukodystrophy heterozygotes. Metab. Brain Dis. 2015, 30, 1279–1284. [Google Scholar] [CrossRef]
- Berger, J.; Forss-Petter, S.; Eichler, F.S. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 2014, 98, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Vinken, P.J.; Bruyn, C.W.; Moser, H.W. X-linked adrenoleukodystrophy. In Handbook Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 1996; Volume 66, pp. 447–483. [Google Scholar]
- Kemp, S.; Berger, J.; Aubourg, P. X-linked adrenoleukodystrophy: Clinical, metabolic, genetic and pathophysiological aspects. Biochim. Biophys. Acta 2012, 1822, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.P.; Moser, H.W.; Saxena, K.M.; Marmion, L.C. Adrenoleukodystrophy: Clinical and biochemical manifestations in carriers. Neurology 1984, 34, 798–801. [Google Scholar] [CrossRef]
- Moser, H.W.; Moser, A.B.; Naidu, S.; Bergin, A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev. Neurosci. 1991, 13, 254–261. [Google Scholar] [CrossRef]
- Van Geel, B.M. Draagsterschap van X-gebonden adrenoleukodystrofie [Carrier state of x-linked adrenoleukodystrophy]. Ned. Tijdschr. Geneeskd. 2000, 144, 1764–1768. (In Dutch) [Google Scholar] [PubMed]
- Schmidt, S.; Träber, F.; Block, W.; Keller, E.; Pohl, C.; von Oertzen, J.; Schild, H.; Schlegel, U.; Klockgether, T. Phenotype assignment in symptomatic female carriers of X-linked adrenoleukodystrophy. J. Neurol. 2001, 248, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Vasco, G.; Aiello, C.; Rizzo, C.; Sancesario, A.; Romano, A.; Favetta, M.; Petrarca, M.; Paone, L.; Castelli, E.; et al. Natural history of a cohort of ABCD1 variant female carriers. Eur. J. Neurol. 2019, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, S.S.; Naidu, S.; Blevins, L.S.; Ladenson, P.W. Assessment of adrenal function in women heterozygous for adrenoleukodystrophy. J. Clin. Endocrinol. Metab. 1997, 82, 856–860. [Google Scholar] [CrossRef]
- Pilz, P.; Schiener, P. Kombination von Morbus Addison und Morbus Schilder bei einer 43 jährigen Frau [Combination of Addison’s and Schilder’s disease in a woman aged 43 years (author’s transl)]. Acta Neuropathol. 1973, 26, 357–360. [Google Scholar] [CrossRef]
- Jung, H.H.; Wimplinger, I.; Jung, S.; Landau, K.; Gal, A.; Heppner, F.L. Phenotypes of female adrenoleukodystrophy. Neurology 2007, 68, 960–961. [Google Scholar] [CrossRef]
- Habekost, C.T.; Schestatsky, P.; Torres, V.F.; de Coelho, D.M.; Vargas, C.R.; Torrez, V.; Oses, J.P.; Portela, L.V.; Pereira Fdos, S.; Matte, U.; et al. Neurological impairment among heterozygote women for X-linked Adrenoleukodystrophy: A case control study on a clinical, neurophysiological and biochemical characteristics. Orphanet. J. Rare Dis. 2014, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Azar, C.; Shor, N.; Nadjar, Y. Adrenomyeloneuropathy masquerading as chronic myelitis. JAMA Neurol. 2020, 77, 522–523. [Google Scholar] [CrossRef]
- Kemp, S.; Pujol, A.; Waterham, H.R.; van Geel, B.M.; Boehm, C.D.; Raymond, G.V.; Cutting, G.R.; Wanders, R.J.; Moser, H.W. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: Role in diagnosis and clinical correlations. Hum. Mutat. 2001, 18, 499–515. [Google Scholar] [CrossRef]
- Palakuzhiyil, S.V.; Christopher, R.; Chandra, S.R. Deciphering the modifiers for phenotypic variability of X-linked adrenoleukodystrophy. World J. Biol Chem. 2020, 11, 99–111. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lachtermacher, M.B.; Seuánez, H.N.; Moser, A.B.; Moser, H.W.; Smith, K.D. Determination of 30 X-linked adrenoleukodystrophy mutations, including 15 not previously described. Hum. Mutat. 2000, 15, 348–353. [Google Scholar] [CrossRef]
- Coll, M.J.; Palau, N.; Camps, C.; Ruiz, M.; Pàmpols, T.; Girós, M. X-linked adrenoleukodystrophy in Spain. Identification of 26 novel mutations in the ABCD1 gene in 80 patients. Improvement of genetic counseling in 162 relative females. Clin. Genet. 2005, 67, 418–424. [Google Scholar] [CrossRef]
- Asheuer, M.; Bieche, I.; Laurendeau, I.; Moser, A.; Hainque, B.; Vidaud, M.; Aubourg, P. Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2005, 14, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Montagna, G.; Di Biase, A.; Cappa, M.; Melone, M.A.; Piantadosi, C.; Colabianchi, D.; Patrono, C.; Attori, L.; Cannelli, N.; Cotrufo, R.; et al. Identification of seven novel mutations in ABCD1 by a DHPLC-based assay in Italian patients with X-linked adrenoleukodystrophy. Hum. Mutat. 2005, 25, 222. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xiong, H.; Wu, Y.; Zhang, Y.H.; Bao, X.H.; Jiang, Y.W.; Wu, X.R. ABCD1 gene mutations in Chinese patients with X-linked adrenoleukodystrophy. Pediatr. Neurol. 2005, 33, 114–120. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Sakai, N.; Hamada, Y.; Tachibana, M.; Hasegawa, Y.; Kiyohara, Y.; Yamada, H.; Murakami, M.; Kondou, H.; Kimura-Ohba, S.; et al. Clinical aspects and adrenal functions in eleven Japanese children with X-linked adrenoleukodystrophy. Endocr. J. 2010, 57, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Shimozawa, N.; Honda, A.; Kajiwara, N.; Kozawa, S.; Nagase, T.; Takemoto, Y.; Suzuki, Y. X-linked adrenoleukodystrophy: Diagnostic and follow-up system in Japan. J. Hum. Genet. 2011, 56, 106–109. [Google Scholar] [CrossRef]
- Pereira Fdos, S.; Matte, U.; Habekost, C.T.; de Castilhos, R.M.; El Husny, A.S.; Lourenço, C.M.; Vianna-Morgante, A.M.; Giuliani, L.; Galera, M.F.; Honjo, R.; et al. Mutations, clinical findings and survival estimates in South American patients with X-linked adrenoleukodystrophy. PLoS ONE 2012, 7, e34195. [Google Scholar] [CrossRef]
- Zhan, Z.X.; Liao, X.X.; Du, J.; Luo, Y.Y.; Hu, Z.T.; Wang, J.L.; Yan, X.X.; Zhang, J.G.; Dai, M.Z.; Zhang, P.; et al. Exome sequencing released a case of X-linked adrenoleukodystrophy mimicking recessive hereditary spastic paraplegia. Eur. J. Med. Genet. 2013, 56, 375–378. [Google Scholar] [CrossRef]
- Niu, Y.F.; Ni, W.; Wu, Z.Y. ABCD1 mutations and phenotype distribution in Chinese patients with X-linked adrenoleukodystrophy. Gene 2013, 522, 117–120. [Google Scholar] [CrossRef]
- Park, H.J.; Shin, H.Y.; Kang, H.C.; Choi, B.O.; Suh, B.C.; Kim, H.J.; Choi, Y.C.; Lee, P.H.; Kim, S.M. Clinical and genetic aspects in twelve Korean patients with adrenomyeloneuropathy. Yonsei Med. J. 2014, 55, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.Y.; Cai, Y.N.; Liang, C.L.; Peng, M.Z.; Sheng, H.Y.; Fan, L.P.; Lin, R.Z.; Jiang, H.; Huang, Y.; Liu, L. Clinical, biochemical, neuroimaging and molecular findings of X-linked Adrenoleukodystrophy patients in South China. Metab. Brain Dis. 2015, 30, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Ping, L.L.; Bao, X.H.; Wang, A.H.; Pan, H.; Wu, Y.; Xiong, H.; Zhang, Y.H.; Jiang, Y.W.; Qin, J.; Wu, X.R. The genotype and phenotype studies of 40 Chinese patients with X-linked adrenoleukodystrophy (X-ALD). Beijing Da Xue Xue Bao Yi Xue Ban. 2006, 38, 66–70. [Google Scholar]
- Maestri, N.E.; Beaty, T.H. Predictions of a 2-locus model for disease heterogeneity: Application to adrenoleukodystrophy. Am. J. Med. Genet. 1992, 44, 576–582. [Google Scholar] [CrossRef]
- Migeon, B.R.; Moser, H.W.; Moser, A.B.; Axelman, J.; Sillence, D.; Norum, R.A. Adrenoleukodystrophy: Evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc. Natl. Acad. Sci. USA 1981, 78, 5066–5070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margoni, M.; Soli, F.; Sangalli, A.; Bellizzi, M.; Cecchini, E.; Buganza, M. A novel mutation in ABCD1 unveils different clinical phenotypes in a family with adrenoleukodystrophy. J. Clin. Neurosci. 2017, 43, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Paláu-Hernández, S.; Rodriguez-Leyva, I.; Shiguetomi-Medina, J.M. Late onset adrenoleukodystrophy: A review related clinical case report. eNeurologicalSci. J. 2019, 14, 62–67. [Google Scholar] [CrossRef] [PubMed]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 


{kind=link}
{kind=link}