
De Novo Transcriptome Meta-Assembly of the Mixotrophic Freshwater Microalga Euglena gracilis

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.1.1. Public Repositories
2.1.2. In-House Experiments, Cell Culture and Sequencing
2.2. Data Assembly
2.2.1. Data Pre-Processing
2.2.2. Transcriptome Assembly
2.2.3. Transcriptome Decontamination
2.2.4. Generation of a Consensus Transcriptome
2.3. Assessment of Transcriptome Quality
2.4. Transcript Annotation
2.5. Taxonomic Analyses
2.6. Expression Quantification
2.7. Gene Clustering Based on Expression Profiles
2.8. Gene Ontology (Enrichment) Analyses
3. Results and Discussion
3.1. Data Collection/Datasets
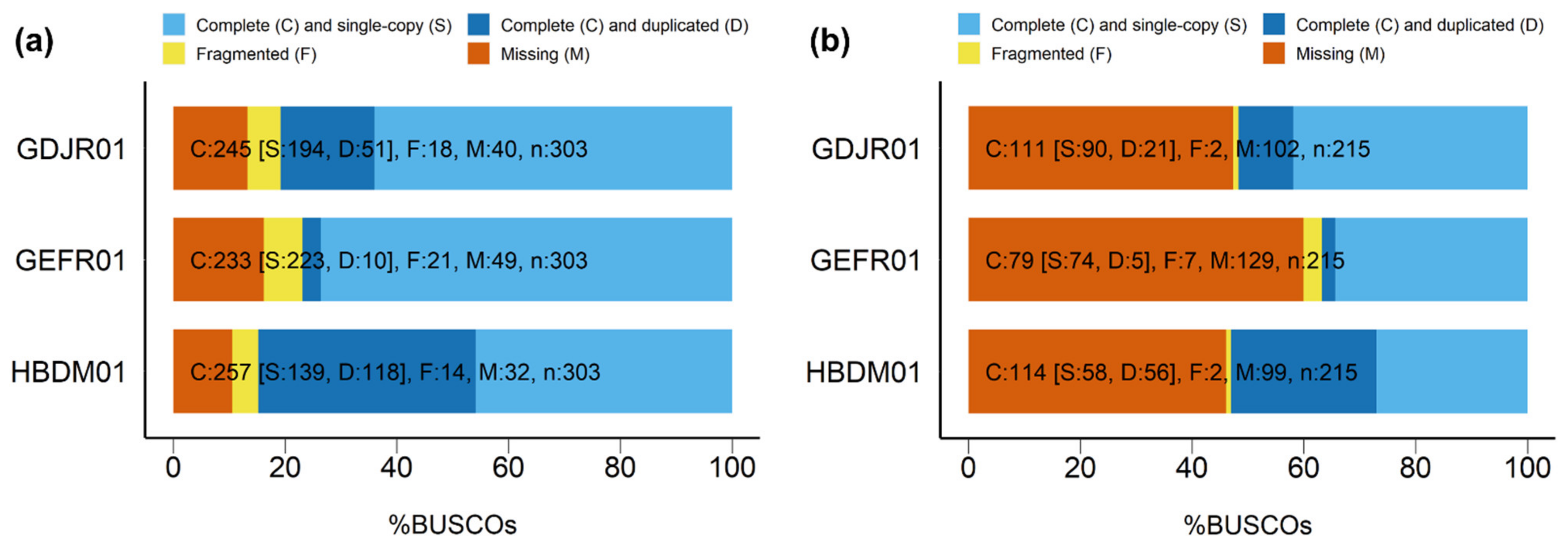
3.2. De Novo Assembly Evaluation
3.2.1. Individual Assemblies
3.2.2. Final Consensus Transcriptome
3.3. Global (Transcriptome) Annotation
3.3.1. Functional Annotation of Transcripts
3.3.2. Taxonomic Annotation of Transcripts
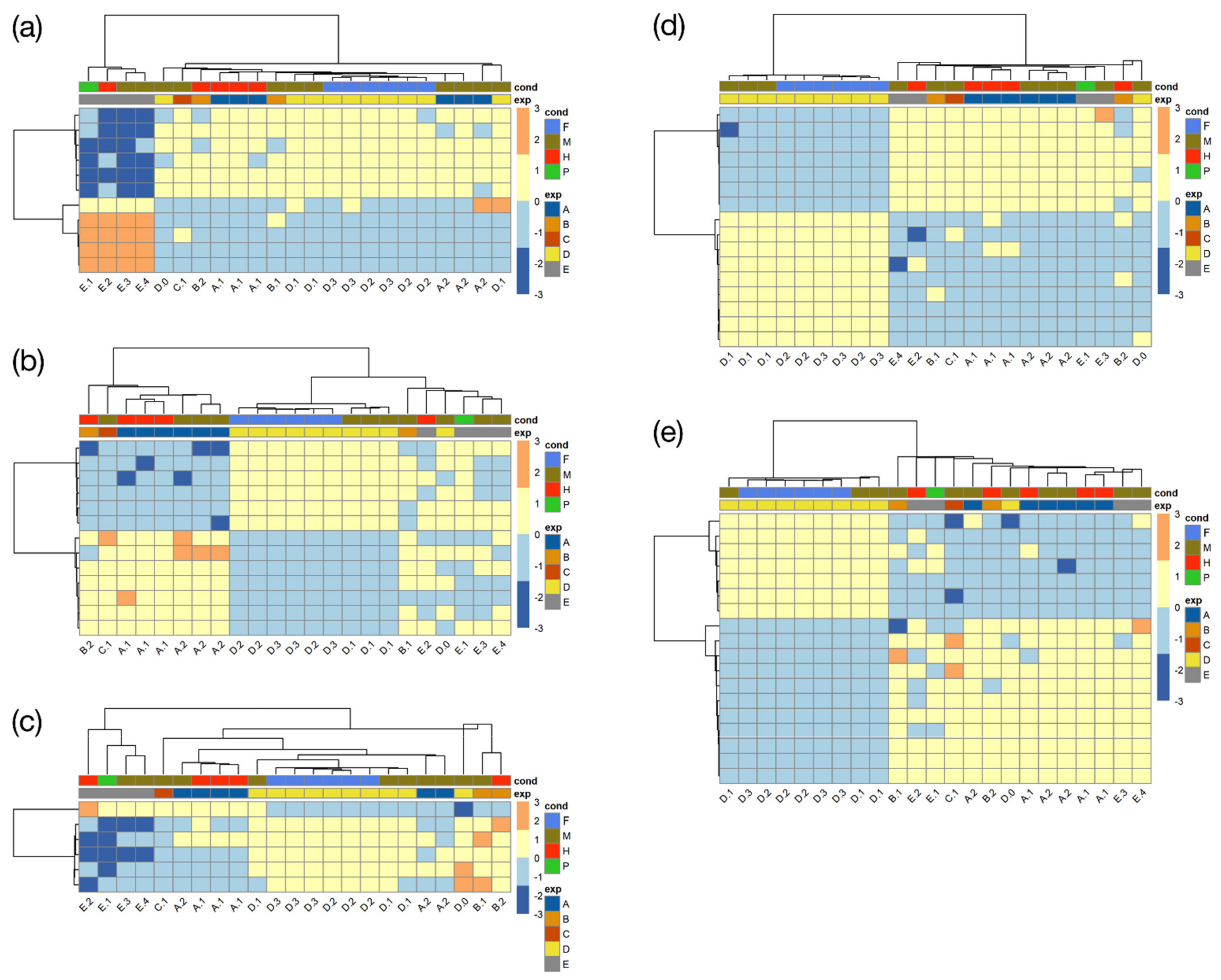
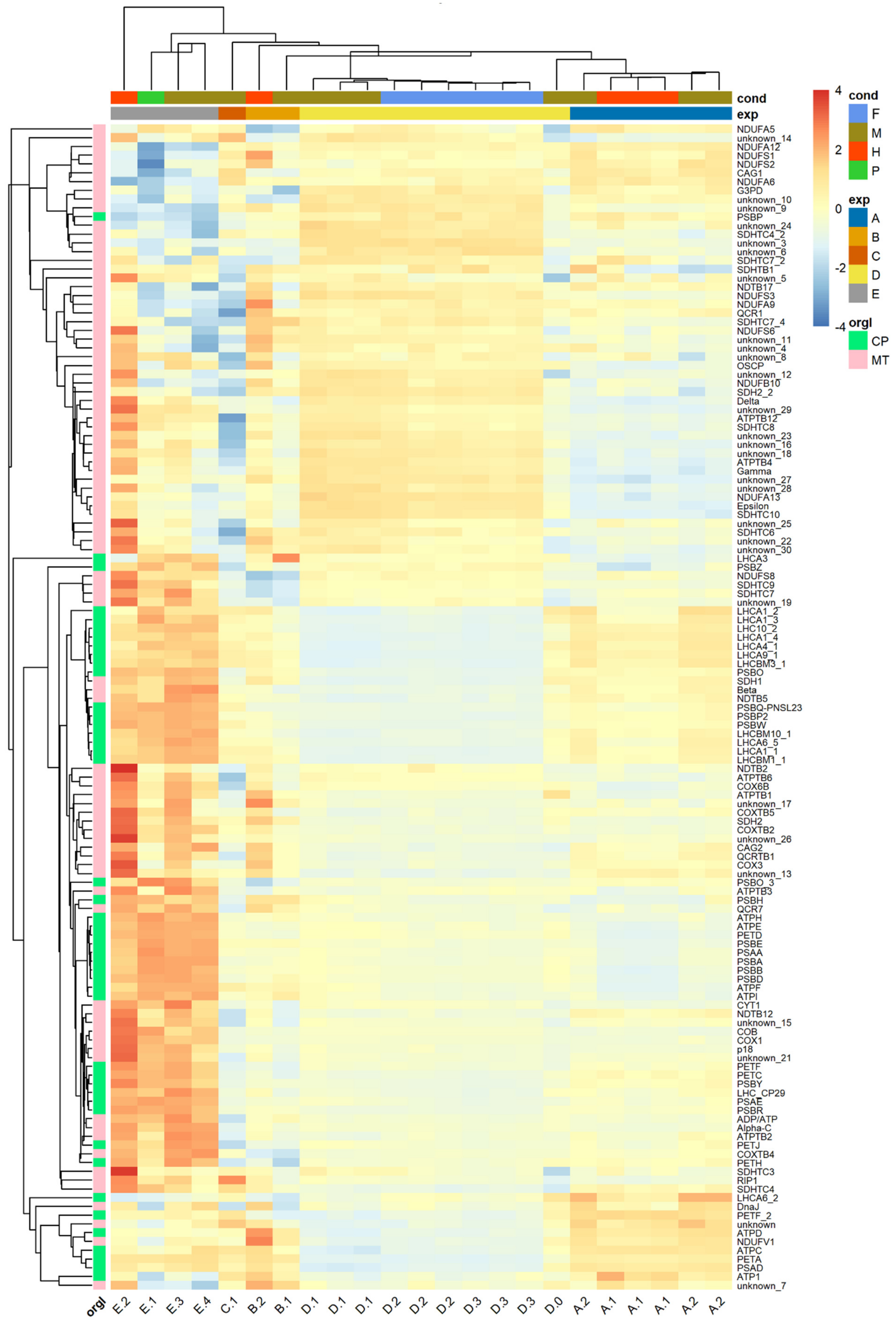
3.4. Systematic Functional Annotation of Top Differentially Expressed Genes
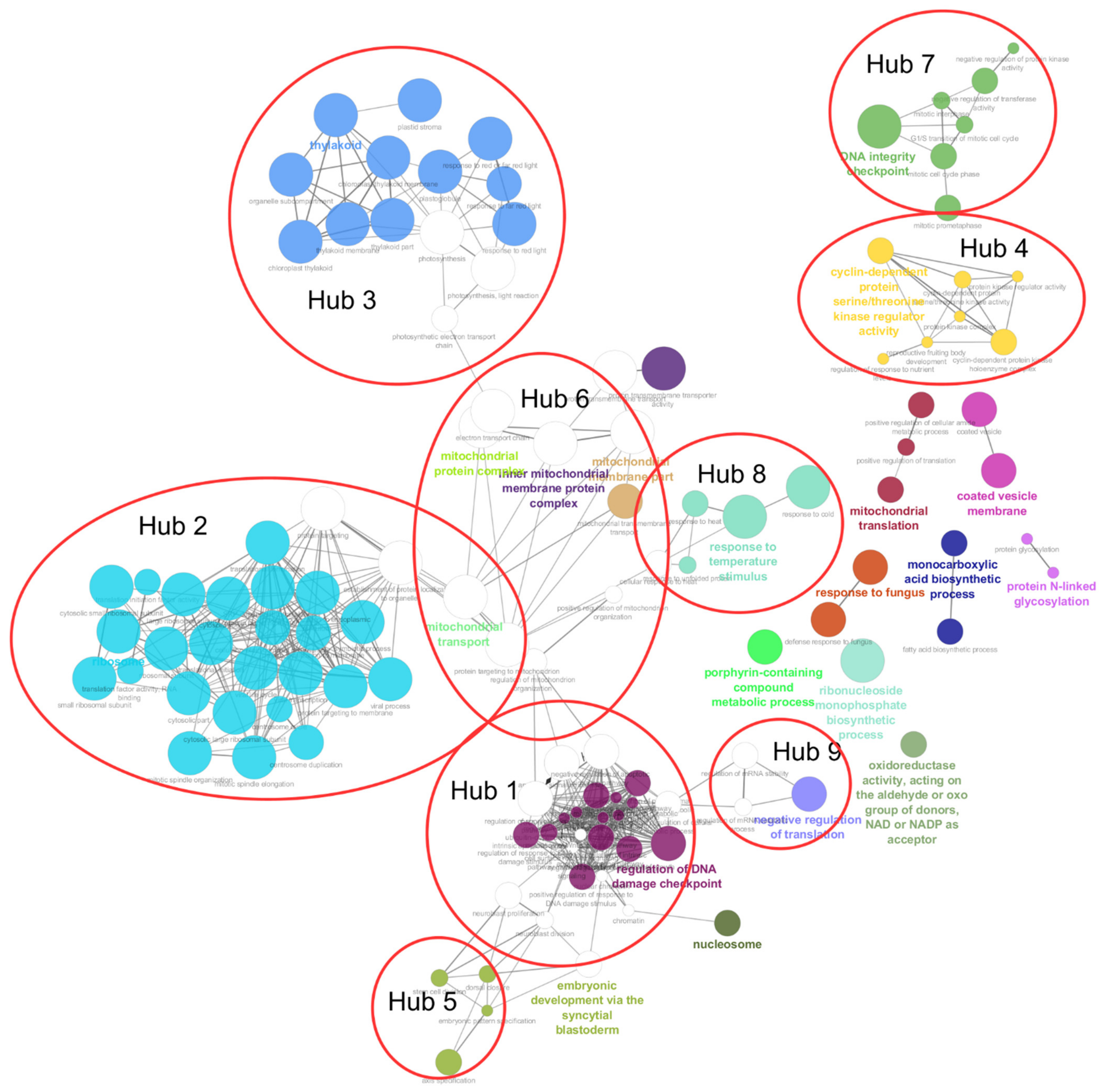
3.5. Cluster Annotation Enrichment Analysis and Gene Co-Expression
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leander, B.S.; Farmer, M.A. Comparative morphology of the euglenid pellicle. II. Diversity of strip substructure. J. Eukaryot. Microbiol. 2001, 48, 202–217. [Google Scholar] [CrossRef]
- Adl, S.M.; Simpson, A.G.; Lane, C.E.; Lukes, J.; Bass, D.; Bowser, S.S.; Brown, M.W.; Burki, F.; Dunthorn, M.; Hampl, V.; et al. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 2012, 59, 429–493. [Google Scholar] [CrossRef] [Green Version]
- Breglia, S.A.; Yubuki, N.; Hoppenrath, M.; Leander, B.S. Ultrastructure and molecular phylogenetic position of a novel euglenozoan with extrusive episymbiotic bacteria: Bihospites bacati n. gen. et sp. (Symbiontida). BMC Microbiol. 2010, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Burki, F. The Eukaryotic Tree of Life from a Global Phylogenomic Perspective. Cold Spring Harb. Perspect. Biol. 2014, 6, a016147. [Google Scholar] [CrossRef] [PubMed]
- Zakrys, B.; Milanowski, R.; Karnkowska, A. Evolutionary Origin of Euglena. Adv. Exp. Med. Biol. 2017, 979, 3–17. [Google Scholar] [CrossRef]
- Butenko, A.; Opperdoes, F.R.; Flegontova, O.; Horak, A.; Hampl, V.; Keeling, P.; Gawryluk, R.M.R.; Tikhonenkov, D.; Flegontov, P.; Lukes, J. Evolution of metabolic capabilities and molecular features of diplonemids, kinetoplastids, and euglenids. BMC Biol. 2020, 18, 23. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, T.E.; Zoltner, M.; Burrell, A.; Nenarokova, A.; Novak Vanclova, A.M.G.; Prasad, B.; Soukal, P.; Santana-Molina, C.; O’Neill, E.; Nankissoor, N.N.; et al. Transcriptome, proteome and draft genome of Euglena gracilis. BMC Biol. 2019, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Vesteg, M.; Hadariova, L.; Horvath, A.; Estrano, C.E.; Schwartzbach, S.D.; Krajcovic, J. Comparative molecular cell biology of phototrophic euglenids and parasitic trypanosomatids sheds light on the ancestor of Euglenozoa. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1701–1721. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.; Lapaille, M.; Degand, H.; Cilibrasi, L.; Villavicencio-Queijeiro, A.; Morsomme, P.; Gonzalez-Halphen, D.; Field, M.C.; Remacle, C.; Baurain, D.; et al. The mitochondrial respiratory chain of the secondary green alga Euglena gracilis shares many additional subunits with parasitic Trypanosomatidae. Mitochondrion 2014, 19 Pt B, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.P.; Quail, M.A.; Berriman, M. Insights into the genome sequence of a free-living Kinetoplastid: Bodo saltans (Kinetoplastida: Euglenozoa). BMC Genom. 2008, 9, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschamps, P.; Lara, E.; Marande, W.; Lopez-Garcia, P.; Ekelund, F.; Moreira, D. Phylogenomic analysis of kinetoplastids supports that trypanosomatids arose from within bodonids. Mol. Biol. Evol. 2011, 28, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Cavalier-Smith, T. Eukaryote kingdoms: Seven or nine? Biosystems 1981, 14, 461–481. [Google Scholar] [CrossRef]
- Sogin, M.L.; Elwood, H.J.; Gunderson, J.H. Evolutionary diversity of eukaryotic small-subunit rRNA genes. Proc. Natl. Acad. Sci. USA 1986, 83, 1383–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier, L.H.; Keller, M.; Chan, R.L.; Fournier, R.; Weil, J.H.; Imbault, P. Short leader sequences may be transferred from small RNAs to pre-mature mRNAs by trans-splicing in Euglena. EMBO J. 1991, 10, 2621–2625. [Google Scholar] [CrossRef] [PubMed]
- Montrichard, F.; Le Guen, F.; Laval-Martin, D.L.; Davioud-Charvet, E. Evidence for the co-existence of glutathione reductase and trypanothione reductase in the non-trypanosomatid Euglenozoa: Euglena gracilis Z. FEBS Lett. 1999, 442, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Sibbald, S.J.; Archibald, J.M. Genomic Insights into Plastid Evolution. Genome Biol. Evol. 2020, 12, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Gilson, P.R.; Su, V.; McFadden, G.I.; Keeling, P.J. The complete chloroplast genome of the chlorarachniophyte Bigelowiella natans: Evidence for independent origins of chlorarachniophyte and euglenid secondary endosymbionts. Mol. Biol. Evol. 2007, 24, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turmel, M.; Gagnon, M.C.; O’Kelly, C.J.; Otis, C.; Lemieux, C. The chloroplast genomes of the green algae Pyramimonas, Monomastix, and Pycnococcus shed new light on the evolutionary history of prasinophytes and the origin of the secondary chloroplasts of euglenids. Mol. Biol. Evol. 2009, 26, 631–648. [Google Scholar] [CrossRef]
- Jackson, C.; Knoll, A.H.; Chan, C.X.; Verbruggen, H. Plastid phylogenomics with broad taxon sampling further elucidates the distinct evolutionary origins and timing of secondary green plastids. Sci. Rep. 2018, 8, 1523. [Google Scholar] [CrossRef]
- Gibbs, S.P. The chloroplasts of Euglena may have evolved from symbiotic green algae. Can. J. Bot. 1978, 56, 2883–2889. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Membrane heredity and early chloroplast evolution. Trends Plant Sci. 2000, 5, 174–182. [Google Scholar] [CrossRef]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Larkum, A.W.; Lockhart, P.J.; Howe, C.J. Shopping for plastids. Trends Plant. Sci. 2007, 12, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.J.; Barbrook, A.C.; Nisbet, R.E.; Lockhart, P.J.; Larkum, A.W. The origin of plastids. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J. The number, speed, and impact of plastid endosymbioses in eukaryotic evolution. Annu. Rev. Plant Biol. 2013, 64, 583–607. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Toledo, R.I.; Lopez-Garcia, P.; Moreira, D. Horizontal and endosymbiotic gene transfer in early plastid evolution. New Phytol. 2019, 224, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Teich, R.; Zauner, S.; Baurain, D.; Brinkmann, H.; Petersen, J. Origin and distribution of Calvin cycle fructose and sedoheptulose bisphosphatases in plantae and complex algae: A single secondary origin of complex red plastids and subsequent propagation via tertiary endosymbioses. Protist 2007, 158, 263–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, J.; Ludewig, A.K.; Michael, V.; Bunk, B.; Jarek, M.; Baurain, D.; Brinkmann, H. Chromera velia, endosymbioses and the rhodoplex hypothesis--plastid evolution in cryptophytes, alveolates, stramenopiles, and haptophytes (CASH lineages). Genome Biol. Evol. 2014, 6, 666–684. [Google Scholar] [CrossRef] [Green Version]
- Barbrook, A.C.; Howe, C.J.; Purton, S. Why are plastid genomes retained in non-photosynthetic organisms? Trends Plant Sci. 2006, 11, 101–108. [Google Scholar] [CrossRef]
- Singer, A.; Poschmann, G.; Muhlich, C.; Valadez-Cano, C.; Hansch, S.; Huren, V.; Rensing, S.A.; Stuhler, K.; Nowack, E.C.M. Massive Protein Import into the Early-Evolutionary-Stage Photosynthetic Organelle of the Amoeba Paulinella chromatophora. Curr. Biol. 2017, 27, 2763–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadinejad, N.; Dagan, T.; Martin, W. Genome history in the symbiotic hybrid Euglena gracilis. Gene 2007, 402, 35–39. [Google Scholar] [CrossRef]
- Maruyama, S.; Suzaki, T.; Weber, A.P.; Archibald, J.M.; Nozaki, H. Eukaryote-to-eukaryote gene transfer gives rise to genome mosaicism in euglenids. BMC Evol. Biol. 2011, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, M.; Myers, J. Growth and photosynthetic characteristics of Euglena gracilis. Archiv. Für Mikrobiol. 1952, 17, 384–402. [Google Scholar] [CrossRef]
- Wilson, B.W.; Danforth, W.F. The extent of acetate and ethanol oxidation by Euglena gracilis. J. Gen. Microbiol. 1958, 18, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Buetow, D. Ethanol stimulation of oxidative metabolism in Euglena gracilis. Nature 1961, 190, 1196. [Google Scholar] [CrossRef]
- Mego, J.L.; Farb, R.M. Alcohol dehydrogenases of Euglena gracilis, strain Z. Biochim. Biophys. Acta 1974, 350, 237–239. [Google Scholar] [CrossRef]
- Sharpless, T.K.; Butow, R.A. An inducible alternate terminal oxidase in Euglena gracilis mitochondria. J. Biol. Chem. 1970, 245, 58–70. [Google Scholar] [CrossRef]
- App, A.A.; Jagendorf, A.T. Repression of chloroplast development in Euglena gracilis by substrates. J. Protozool. 1963, 10, 340–343. [Google Scholar] [CrossRef]
- Buetow, D.E. Acetate repression of chlorophyll synthesis in Euglena gracilis. Nature 1967, 213, 1127–1128. [Google Scholar] [CrossRef]
- Vannini, G.L. Degeneration and regeneration of chloroplasts in Euglena gracilis grown in the presence of acetate: Ultrastructural evidence. J. Cell Sci. 1983, 61, 413–422. [Google Scholar] [CrossRef]
- Calvayrac, R.; Laval-Martin, D.; Briand, J.; Farineau, J. Paramylon synthesis by Euglena gracilis photoheterotrophically grown under low O2 pressure: Description of a mitochloroplast complex. Planta 1981, 153, 6–13. [Google Scholar] [CrossRef]
- Monfils, A.K.; Triemer, R.E.; Bellairs, E.F. Characterization of paramylon morphological diversity in photosynthetic euglenoids (Euglenales, Euglenophyta). Phycologia 2011, 50, 156–169. [Google Scholar] [CrossRef]
- Shibakami, M.; Tsubouchi, G.; Hayashi, M. Thermoplasticization of euglenoid beta-1,3-glucans by mixed esterification. Carbohydr. Polym. 2014, 105, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Shimada, R.; Matsuyama, A.; Yuasa, M.; Sawamura, H.; Yoshida, E.; Suzuki, K. Antitumor activity of the beta-glucan paramylon from Euglena against preneoplastic colonic aberrant crypt foci in mice. Food Funct. 2013, 4, 1685–1690. [Google Scholar] [CrossRef]
- Matsuda, F.; Hayashi, M.; Kondo, A. Comparative profiling analysis of central metabolites in Euglena gracilis under various cultivation conditions. Biosci. Biotechnol. Biochem. 2011, 75, 2253–2256. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, T.; Ogawa, T.; Nakai, R.; Nakazawa, M.; Okazawa, A.; Padermschoke, A.; Nishio, K.; Hirai, M.Y.; Arita, M.; Ohta, D. Wax ester and lipophilic compound profiling of Euglena gracilis by gas chromatography-mass spectrometry: Toward understanding of wax ester fermentation under hypoxia. Metabolomics 2015, 11, 175–183. [Google Scholar] [CrossRef]
- Inui, H.; Ishikawa, T.; Tamoi, M. Wax Ester Fermentation and Its Application for Biofuel Production. Adv. Exp. Med. Biol. 2017, 979, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Ogbonna, J.C.; Tomiyamal, S.; Tanaka, H. Heterotrophic cultivation of Euglena gracilis Z for efficient production of α-tocopherol. J. Appl. Phycol. 1998, 10, 67–74. [Google Scholar] [CrossRef]
- Hallick, R.B.; Hong, L.; Drager, R.G.; Favreau, M.R.; Monfort, A.; Orsat, B.; Spielmann, A.; Stutz, E. Complete sequence of Euglena gracilis chloroplast DNA. Nucleic Acids Res. 1993, 21, 3537–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, D.F.; Gray, M.W. Ribosomal RNA genes in Euglena gracilis mitochondrial DNA: Fragmented genes in a seemingly fragmented genome. Mol. Genet. Genom. 2011, 285, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Dobakova, E.; Flegontov, P.; Skalicky, T.; Lukes, J. Unexpectedly streamlined mitochondrial genome of the euglenozoan Euglena gracilis. Genome Biol. Evol. 2015. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, E.C.; Trick, M.; Hill, L.; Rejzek, M.; Dusi, R.G.; Hamilton, C.J.; Zimba, P.V.; Henrissat, B.; Field, R.A. The transcriptome of Euglena gracilis reveals unexpected metabolic capabilities for carbohydrate and natural product biochemistry. Mol. Biosyst. 2015. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Tomiyama, T.; Maruta, T.; Tomita, M.; Ishikawa, T.; Arakawa, K. De novo assembly and comparative transcriptome analysis of Euglena gracilis in response to anaerobic conditions. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebenezer, T.E.; Carrington, M.; Lebert, M.; Kelly, S.; Field, M.C. Euglena gracilis Genome and Transcriptome: Organelles, Nuclear Genome Assembly Strategies and Initial Features. Adv. Exp. Med. Biol. 2017, 979, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Loppes, R.; Radoux, M. Identification of short promoter regions involved in the transcriptional expression of the nitrate reductase gene in Chlamydomonas reinhardtii. Plant Mol. Biol. 2001, 45, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Gilbert, D. Gene-omes built from mRNA-seq not genome DNA. F1000Research 2016, 5, 1695. [Google Scholar]
- Gilbert, D.G. Genes of the pig, Sus scrofa, reconstructed with EvidentialGene. PeerJ 2019, 7, e6374. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, B.; Fillmore, N.; Bai, Y.; Collins, M.; Thomson, J.A.; Stewart, R.; Dewey, C.N. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014, 15, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Yadav, K.N.S.; Miranda-Astudillo, H.V.; Colina-Tenorio, L.; Bouillenne, F.; Degand, H.; Morsomme, P.; Gonzalez-Halphen, D.; Boekema, E.J.; Cardol, P. Atypical composition and structure of the mitochondrial dimeric ATP synthase from Euglena gracilis. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 267–275. [Google Scholar] [CrossRef]
- Miranda-Astudillo, H.V.; Yadav, K.N.S.; Colina-Tenorio, L.; Bouillenne, F.; Degand, H.; Morsomme, P.; Boekema, E.J.; Cardol, P. The atypical subunit composition of respiratory complexes I and IV is associated with original extra structural domains in Euglena gracilis. Sci. Rep. 2018, 8, 9698. [Google Scholar] [CrossRef] [PubMed]
- Sobotka, R.; Esson, H.J.; Konik, P.; Trskova, E.; Moravcova, L.; Horak, A.; Dufkova, P.; Obornik, M. Extensive gain and loss of photosystem I subunits in chromerid algae, photosynthetic relatives of apicomplexans. Sci. Rep. 2017, 7, 13214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koziol, A.G.; Durnford, D.G. Euglena Light-Harvesting Complexes Are Encoded by Multifarious Polyprotein mRNAs that Evolve in Concert. Mol. Biol. Evol. 2008, 25, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vlierberghe, M.; Philippe, H.; Baurain, D. Broadly sampled orthologous groups of eukaryotic proteins for the phylogenetic study of plastid-bearing lineages. BMC Res. Notes 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Cornet, L.; Meunier, L.; Van Vlierberghe, M.; Leonard, R.R.; Durieu, B.; Lara, Y.; Misztak, A.; Sirjacobs, D.; Javaux, E.J.; Philippe, H.; et al. Consensus assessment of the contamination level of publicly available cyanobacterial genomes. PLoS ONE 2018, 13, e0200323. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Leek, J.T. Svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions, R Package Version 2.0.7-1. 2018. Available online: https://cran.r-project.org/web/packages/cluster/index.html (accessed on 28 May 2021).
- Dunn, J.C. Well-separated clusters and optimal fuzzy partitions. J. Cybern. 1974, 4, 95–104. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Pheatmap: Pretty Heatmaps, R Package Version 1.0.12. 2019. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 28 May 2021).
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Lusk, R.W. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data. PLoS ONE 2014, 9, e110808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.J.; Xu, G.; Morici, L.; Splinter Bon-Durant, S.; Baddoo, M.; Lin, Z.; Fewell, C.; Taylor, C.M.; Flemington, E.K. Microbial contamination in next generation sequencing: Implications for sequence-based analysis of clinical samples. PLoS Pathog. 2014, 10, e1004437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simion, P.; Philippe, H.; Baurain, D.; Jager, M.; Richter, D.J.; Di Franco, A.; Roure, B.; Satoh, N.; Queinnec, E.; Ereskovsky, A.; et al. A Large and Consistent Phylogenomic Dataset Supports Sponges as the Sister Group to All Other Animals. Curr. Biol. 2017, 27, 958–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tae, H.; Karunasena, E.; Bavarva, J.H.; McIver, L.J.; Garner, H.R. Large scale comparison of non-human sequences in human sequencing data. Genomics 2014, 104, 453–458. [Google Scholar] [CrossRef]
- Nakazawa, M.; Minami, T.; Teramura, K.; Kumamoto, S.; Hanato, S.; Takenaka, S.; Ueda, M.; Inui, H.; Nakano, Y.; Miyatake, K. Molecular characterization of a bifunctional glyoxylate cycle enzyme, malate synthase/isocitrate lyase, in Euglena gracilis. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2005, 141, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Nishimura, M.; Inoue, K.; Ueda, M.; Inui, H.; Nakano, Y.; Miyatake, K. Characterization of a bifunctional glyoxylate cycle enzyme, malate synthase/isocitrate lyase, of Euglena gracilis. J. Eukaryot. Microbiol. 2011, 58, 128–133. [Google Scholar] [CrossRef]
- Liu, F.; Thatcher, J.D.; Barral, J.M.; Epstein, H.F. Bifunctional glyoxylate cycle protein of Caenorhabditis elegans: A developmentally regulated protein of intestine and muscle. Dev. Biol. 1995, 169, 399–414. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, D. Evigene Versus Transdecoder for Proteins from Transcripts. Available online: https://sourceforge.net/p/evidentialgene/blog/2017/11/-evigene-versus-transdecoder-for-proteins-from-transcripts/ (accessed on 3 April 2021).
- Kitaoka, M.; Sasaki, T.; Taniguchi, H. Purification and properties of laminaribiose phosphorylase (EC 2.4 1.31) from Euglena gracilis Z. Arch. Biochem. Biophys. 1993, 304, 508–514. [Google Scholar] [CrossRef]
- Kuhaudomlarp, S.; Patron, N.J.; Henrissat, B.; Rejzek, M.; Saalbach, G.; Field, R.A. Identification of Euglena gracilis beta-1,3-glucan phosphorylase and establishment of a new glycoside hydrolase (GH) family GH149. J. Biol. Chem. 2018, 293, 2865–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsanti, L.; Evangelista, V.; Passarelli, V.; Frassanito, A.M.; Gualtieri, P. Fundamental questions and concepts about photoreception and the case of Euglena gracilis. Integr. Biol. 2012, 4, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Gallo, J.M.; Schrevel, J. Homologies between paraflagellar rod proteins from trypanosomes and euglenoids revealed by a monoclonal antibody. Eur. J. Cell Biol. 1985, 36, 163–168. [Google Scholar] [PubMed]
- Cachon, J.; Cachon, M.; Cosson, M.-P.; Cosson, J. The paraflagellar rod: A structure in search of a function. Biol. Cell 1988, 63, 169–181. [Google Scholar] [CrossRef]
- Maharana, B.R.; Tewari, A.K.; Singh, V. An overview on kinetoplastid paraflagellar rod. J. Parasit. Dis. 2015, 39, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verni, F.; Rosati, G.; Lenzi, P.; Barsanti, L.; Passarelli, V.; Gualtieri, P. Morphological relationship between paraflagellar swelling and paraxial rod in Euglena gracilis. Micron Microsc. Acta 1992, 23, 37–44. [Google Scholar] [CrossRef]
- Iseki, M.; Matsunaga, S.; Murakami, A.; Ohno, K.; Shiga, K.; Yoshida, K.; Sugai, M.; Takahashi, T.; Hori, T.; Watanabe, M. A blue-light-activated adenylyl cyclase mediates photoavoidance in Euglena gracilis. Nature 2002, 415, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Koumura, Y.; Suzuki, T.; Yoshikawa, S.; Watanabe, M.; Iseki, M. The origin of photoactivated adenylyl cyclase (PAC), the Euglena blue-light receptor: Phylogenetic analysis of orthologues of PAC subunits from several euglenoids and trypanosome-type adenylyl cyclases from Euglena gracilis. Photochem. Photobiol. Sci. 2004, 3, 580–586. [Google Scholar] [CrossRef]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [Green Version]
- Markunas, C.M.; Triemer, R.E. Evolutionary History of the Enzymes Involved in the Calvin-Benson Cycle in Euglenids. J. Eukaryot. Microbiol. 2016, 63, 326–339. [Google Scholar] [CrossRef]
- Lakey, B.; Triemer, R.; Müller, K. The tetrapyrrole synthesis pathway as a model of horizontal gene transfer in euglenoids. J. Phycol. 2017, 53, 198–217. [Google Scholar] [CrossRef]
- Ponce-Toledo, R.I.; Moreira, D.; Lopez-Garcia, P.; Deschamps, P. Secondary Plastids of Euglenids and Chlorarachniophytes Function with a Mix of Genes of Red and Green Algal Ancestry. Mol. Biol. Evol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Novak Vanclova, A.M.G.; Zoltner, M.; Kelly, S.; Soukal, P.; Zahonova, K.; Fussy, Z.; Ebenezer, T.E.; Lacova Dobakova, E.; Elias, M.; Lukes, J.; et al. Metabolic quirks and the colourful history of the Euglena gracilis secondary plastid. New Phytol. 2020, 225, 1578–1592. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Principles of protein and lipid targeting in secondary symbiogenesis: Euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree. J. Eukaryot. Microbiol. 1999, 46, 347–366. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Genomic reduction and evolution of novel genetic membranes and protein-targeting machinery in eukaryote-eukaryote chimaeras (meta-algae). Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 109–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenci, U.; Bhattacharya, D.; Weber, A.P.M.; Colleoni, C.; Subtil, A.; Ball, S.G. Biotic Host-Pathogen Interactions as Major Drivers of Plastid Endosymbiosis. Trends Plant Sci. 2017, 22, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Curtis, B.A.; Tanifuji, G.; Burki, F.; Gruber, A.; Irimia, M.; Maruyama, S.; Arias, M.C.; Ball, S.G.; Gile, G.H.; Hirakawa, Y.; et al. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 2012, 6, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Read, B.A.; Kegel, J.; Klute, M.J.; Kuo, A.; Lefebvre, S.C.; Maumus, F.; Mayer, C.; Miller, J.; Monier, A.; Salamov, A.; et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 2013, 499, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrell, R.G.; Gile, G.; McCallum, G.; Meheust, R.; Bapteste, E.P.; Klinger, C.M.; Brillet-Gueguen, L.; Freeman, K.D.; Richter, D.J.; Bowler, C. Chimeric origins of ochrophytes and haptophytes revealed through an ancient plastid proteome. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Simion, P.; Belkhir, K.; Francois, C.; Veyssier, J.; Rink, J.C.; Manuel, M.; Philippe, H.; Telford, M.J. A software tool ’CroCo’ detects pervasive cross-species contamination in next generation sequencing data. BMC Biol. 2018, 16, 28. [Google Scholar] [CrossRef]
- Kumar, S.; Jones, M.; Koutsovoulos, G.; Clarke, M.; Blaxter, M. Blobology: Exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 2013, 4, 237. [Google Scholar] [CrossRef] [Green Version]
- Lukes, J.; Leander, B.S.; Keeling, P.J. Cascades of convergent evolution: The corresponding evolutionary histories of euglenozoans and dinoflagellates. Proc. Natl. Acad. Sci. USA 2009, 106 (Suppl. 1), 9963–9970. [Google Scholar] [CrossRef] [Green Version]
- Goh, W.W.B.; Wang, W.; Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Almasi, A.; Pescod, M.B. Wastewater treatment mechanisms in anoxic stabilization ponds. Water Sci. Technol. 1996, 33, 125–132. [Google Scholar] [CrossRef]
- Hauslage, J.; Strauch, S.M.; Eßmann, O.; Haag, F.W.M.; Richter, P.; Krüger, J.; Stoltze, J.; Becker, I.; Nasir, A.; Bornemann, G.; et al. Eu:CROPIS—“Euglena gracilis: Combined Regenerative Organic-food Production in Space”—A Space Experiment Testing Biological Life Support Systems Under Lunar And Martian Gravity. Microgravity Sci. Technol. 2018, 30, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Perez, E. Analyses Biochimiques, Protéomiques et Transcriptomiques du Métabolisme Énergétique chez L’algue Secondaire verte Euglena gracilis (Euglenozoa, Excavata); Université de Liège: Liège, Belgique, 2015. [Google Scholar]
- Simpson, A.G.; Stevens, J.R.; Lukes, J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006, 22, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Schwartzbach, S.D. Photo and nutritional regulation of Euglena organelle development. Euglena Biochem. Cell Mol. Biol. 2017, 159–182. [Google Scholar] [CrossRef]
- Gain, G.; Vega de Luna, F.; Cordoba, J.; Perez, E.; Degand, H.; Morsomme, P.; Thiry, M.; Baurain, D.; Pierangelini, M.; Cardol, P. Trophic state alters the mechanism whereby energetic coupling between photosynthesis and respiration occurs in Euglena gracilis. New Phytol. 2021. (in revision). [Google Scholar]
- Koski, L.B.; Golding, G.B. The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 2001, 52, 540–542. [Google Scholar] [CrossRef]
- Triemer, R.E. Feeding in Peranema trichophorum revisited (Euglenophyta) 1. J. Phycol. 1997, 33, 649–654. [Google Scholar] [CrossRef]
- Schnepf, E.; Deichgräber, G. «Myzocytosis», a kind of endocytosis with implications to compartmentation in endosymbiosis. Naturwissenschaften 1984, 71, 218–219. [Google Scholar] [CrossRef]
- Hehenberger, E.; Gast, R.J.; Keeling, P.J. A kleptoplastidic dinoflagellate and the tipping point between transient and fully integrated plastid endosymbiosis. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodyl, A. Did some red alga-derived plastids evolve via kleptoplastidy? A hypothesis. Biol. Rev. Camb. Philos. Soc. 2018, 93, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Yubuki, N.; Leander, B.S. Morphostasis in a novel eukaryote illuminates the evolutionary transition from phagotrophy to phototrophy: Description of Rapaza viridis n. gen. et sp. (Euglenozoa, Euglenida). BMC Evol. Biol. 2012, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. Code | Study Acc. | Sample Code | Run Acc. | Temp. | Medium | Light | Shaking | Cult Cond. | Harvest Phase | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| A | PRJNA310762 | A.1.1 | SRR3159774 | 25 | R + C | D | 0 | H | Exp | [7] |
| A.1.2 | SRR3159775 | 25 | R + C | D | 0 | H | Exp | |||
| A.1.3 | SRR3159776 | 25 | R + C | D | 0 | H | Exp | |||
| A.2.1 | SRR3159777 | 25 | R + C | LL | 0 | M | Exp | |||
| A.2.2 | SRR3159778 | 25 | R + C | LL | 0 | M | Exp | |||
| A.2.3 | SRR3159779 | 25 | R + C | LL | 0 | M | Exp | |||
| B | PRJEB10085 | B.1 | ERR974915 | 21 | M + C | LL | 0 | M | Stat | [52] |
| B.2 | ERR974916 | 30 | R+C | D | 200 | H | Stat | |||
| C | PRJNA298469 | C.0 | SRR2628535 | 25 | M | LL | 0 | M | Stat | [7] |
| D | PRJNA289402 | D.0 | SRR3195326 | 26 | R+C | HL | 120 | M | Stat | [53] |
| D.1.1 | SRR3195327 | 26 | R+C | HL | 120 | M | Stat | |||
| D.1.2 | SRR3195329 | 26 | R+C | HL | 120 | M | Stat | |||
| D.1.3 | SRR3195331 | 26 | R+C | HL | 120 | M | Stat | |||
| D.2.1 | SRR3195332 | 26 | R+C | HL | 120 | F | Stat | |||
| D.2.2 | SRR3195334 | 26 | R+C | HL | 120 | F | Stat | |||
| D.2.3 | SRR3195335 | 26 | R+C | HL | 120 | F | Stat | |||
| D.3.1 | SRR3195338 | 26 | R+C | HL | 120 | F | Stat | |||
| D.3.2 | SRR3195339 | 26 | R+C | HL | 120 | F | Stat | |||
| D.3.3 | SRR3195340 | 26 | R+C | HL | 120 | F | Stat | |||
| E | PRJEB38787 | E.1 | ERR4227585 | 25 | M | LL | 100 | P | Exp | This study |
| E.2 | ERR4227586 | 25 | M+C | D | 100 | H | Exp | |||
| E.3 | ERR4227587 | 25 | M+C | LL | 100 | M | Exp | |||
| E.4 | ERR4227588 | 25 | M+C | HL | 100 | M | Exp |
| Statistic | A | B | B (Filtered) | C | D | E |
|---|---|---|---|---|---|---|
| ACC | PRJNA310762 | PRJEB10085 | PRJEB10085 | PRJNA298469 | PRJNA289402 | PRJEB38787 |
| REF | [7,52,53] | This study | ||||
| RAW | 61,531,862 | 383,416,636 | 383,416,636 | 27,096,926 | 285,148,782 | 1,902,226,200 |
| PRE | 57,862,467 | 310,302,570 | 310,302,570 | 25,244,887 | 267,779,751 | 875,299,135 |
| CNT | 740 (12 rpm) | 9080 (29 rpm) | 9080 (29 rpm) | 1750 (68 rpm) | 1191 (4 rpm) | 2403 (2 rpm) |
| SEQ | 38,559 | 95,490 | 36,287 | 42,363 | 37,425 | 51,021 |
| MIN | 101 | 101 | 500 | 101 | 101 | 101 |
| MAX | 13,929 | 21,744 | 21,744 | 11,354 | 26,839 | 10,795 |
| MEAN | 1043 | 647 | 1312 | 810 | 1120 | 610 |
| TOTAL | 40,861,413 | 64,426,688 | 47,615,807 | 34,438,742 | 42,382,170 | 31,671,589 |
| SEQ < 200 | 4330 | 17,074 | 0 | 782 | 3051 | 3989 |
| SEQ > 1 k | 16,289 | 18,638 | 18,638 | 10,932 | 17,048 | 7104 |
| SEQ > 10 k | 4 | 15 | 15 | 1 | 13 | 1 |
| ORF | 24,757 | 29,060 | 27,842 | 27,063 | 24,817 | 26,882 |
| ORF (%) | 88% | 82% | 83% | 89% | 87% | 93% |
| N90 | 576 | 347 | 654 | 419 | 606 | 367 |
| N70 | 1140 | 667 | 1101 | 686 | 1187 | 528 |
| N50 | 1607 | 1282 | 1574 | 1014 | 1658 | 753 |
| N30 | 2257 | 2033 | 2243 | 1452 | 2318 | 1090 |
| N10 | 3600 | 4026 | 3707 | 2358 | 3812 | 1850 |
| GC (%) | 64% | 58% | 62% | 64% | 64% | 64% |
| PART | 22,234 | - | 27,730 | 10,129 | 19,663 | 11,602 |
| PART (%) | 24.3% | - | 30.3% | 11.1% | 21.5% | 12.7% |
| Statistic | GEFR01 | GDJR01 | HBDM01 |
|---|---|---|---|
| REF | [7,53] | This study | |
| SEQ | 72,506 | 113,152 | 91,040 1 |
| MIN | 202 | 201 | 201 |
| MAX | 25,763 | 21,553 | 26,839 |
| MEAN | 869 | 1087 | 1096 |
| TOTAL | 63,049,595 | 122,976,775 | 100,187,451 |
| SEQ < 200 | 0 1 | 0 1 | 0 1 |
| SEQ > 1 k | 19,740 | 49,277 | 37,294 |
| SEQ > 10 k | 25 | 27 | 24 |
| ORF 2 | 30,467 | 65,943 | 62,287 |
| ORF (%) | 79% | 73% | 85% |
| N90 | 374 | 523 | 545 |
| N70 | 704 | 1130 | 965 |
| N50 | 1242 | 1604 | 1432 |
| N30 | 1916 | 2181 | 2049 |
| N10 | 3344 | 3347 | 3410 |
| GC (%) | 61% | 63% | 63% |
| CDS | 32,128 | 49,826 | 49,922 |
| GMS-T | 35,929 | 63,432 | 58,542 |
| GMS-T (%) | 49% | 56% | 64% |
| Code | Accession | Reference | GEFR01 | GDJR01 | HBDM01 |
|---|---|---|---|---|---|
| A | PRJNA310762 | [7] | 87.40% | 92.51% | 93.38% |
| B | PRJEB10085 | [52] | 84.68% | 90.13% | 91.49% |
| C | PRJNA298469 | [7] | 80.26% | 91.66% | 90.39% |
| D | PRJNA289402 | [53] | 85.25% | 95.04% | 94.28% |
| E | PRJEB38787 | This study | 75.28% | 51.39% | 80.76% |
| Assembly Score | GEFR01 | GDJR01 | HBDM01 |
|---|---|---|---|
| TransRate Score | 0.1789 | 0.0304 | 0.0430 |
| TransRate Optimal Score | 0.2051 | 0.1729 | 0.0764 |
| Detonate Score | −97,461 × 106 | −97,561 × 106 | −97,459 × 106 |
| Threshold (nt) | Statistic | GEFR01 | GDJR01 | HBDM01 |
|---|---|---|---|---|
| 24 | Forward matches | 24 | 5 | 86 |
| Reverse matches | 21 | 0 | 114 | |
| Total matches | 45 | 5 | 200 | |
| Average length (nt) | 24.00 | 24.00 | 24.00 | |
| 14 | Forward matches | 176 | 16,580 | 3370 |
| Reverse matches | 200 | 12,999 | 3265 | |
| Total matches | 376 | 29,579 | 6635 | |
| Average length (nt) | 16.28 | 15.57 | 15.59 | |
| 12 | Forward matches | 749 | 18,322 | 4403 |
| Reverse matches | 766 | 16,016 | 5397 | |
| Total matches | 1515 | 34,338 | 9800 | |
| Average length (nt) | 13.37 | 15.19 | 14.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordoba, J.; Perez, E.; Van Vlierberghe, M.; Bertrand, A.R.; Lupo, V.; Cardol, P.; Baurain, D. De Novo Transcriptome Meta-Assembly of the Mixotrophic Freshwater Microalga Euglena gracilis. Genes 2021, 12, 842. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060842
Cordoba J, Perez E, Van Vlierberghe M, Bertrand AR, Lupo V, Cardol P, Baurain D. De Novo Transcriptome Meta-Assembly of the Mixotrophic Freshwater Microalga Euglena gracilis. Genes. 2021; 12(6):842. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060842
Chicago/Turabian StyleCordoba, Javier, Emilie Perez, Mick Van Vlierberghe, Amandine R. Bertrand, Valérian Lupo, Pierre Cardol, and Denis Baurain. 2021. "De Novo Transcriptome Meta-Assembly of the Mixotrophic Freshwater Microalga Euglena gracilis" Genes 12, no. 6: 842. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060842