
Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome

 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whole Genome Sequencing
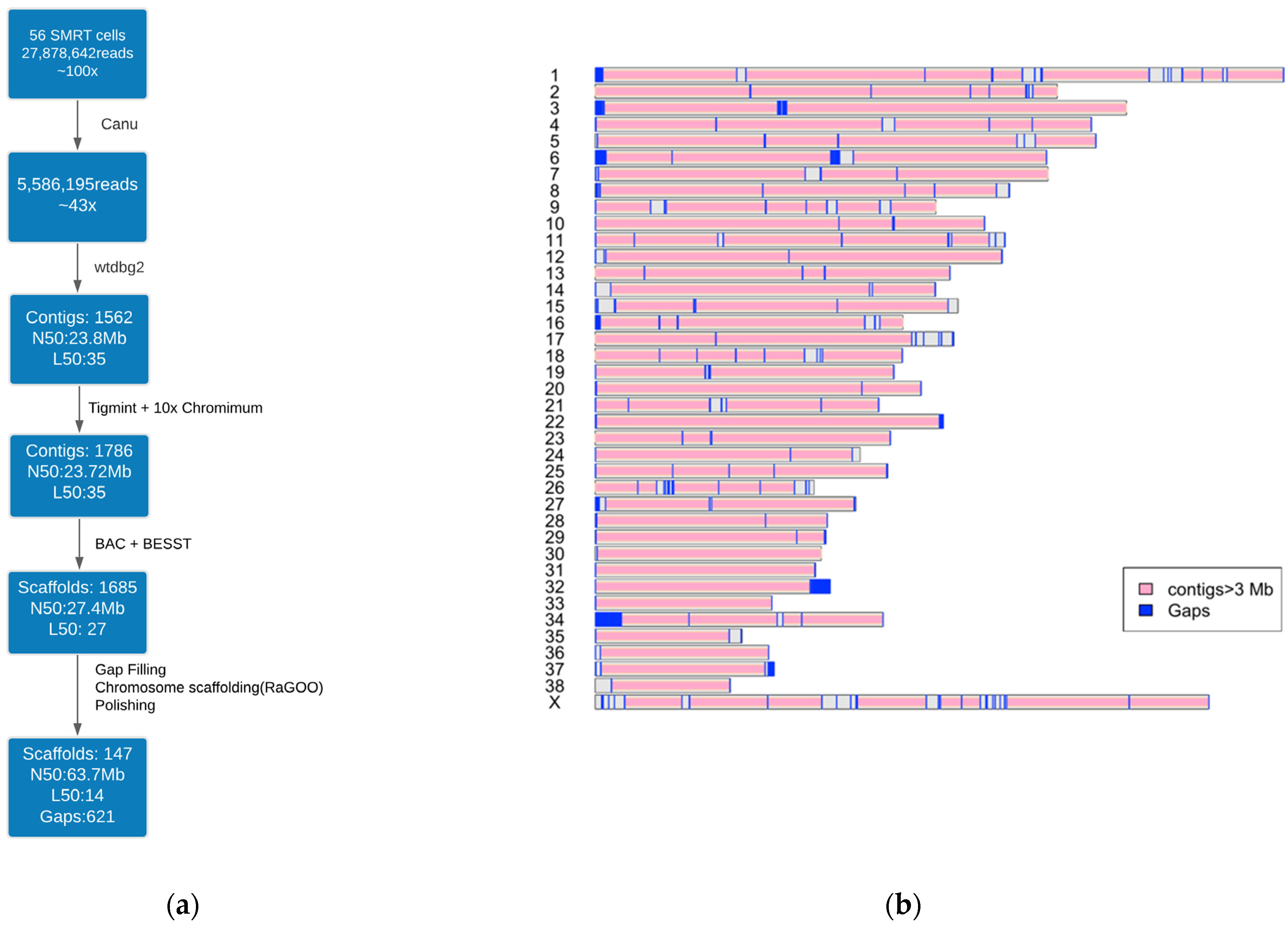
2.2. Genome Assembly Workflow
2.3. Assembly Quality Control
2.3.1. Fosmid End Sequence Alignment
2.3.2. Alignment of Finished BAC Clone Sequences
2.4. Detection of Common Repeats and Segmental Duplications
2.5. Gene Annotation
2.6. Genome Assembly Alignment
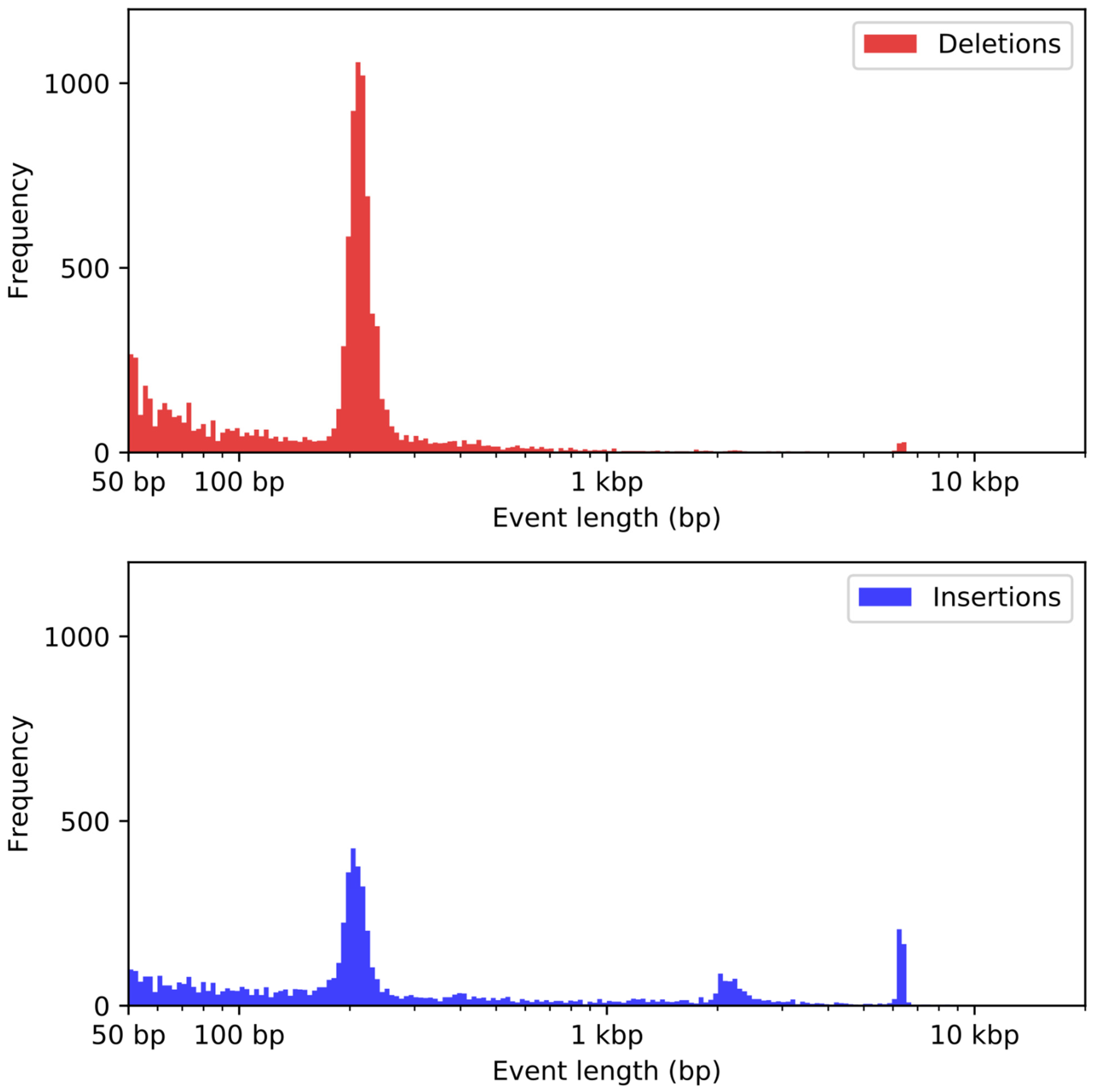
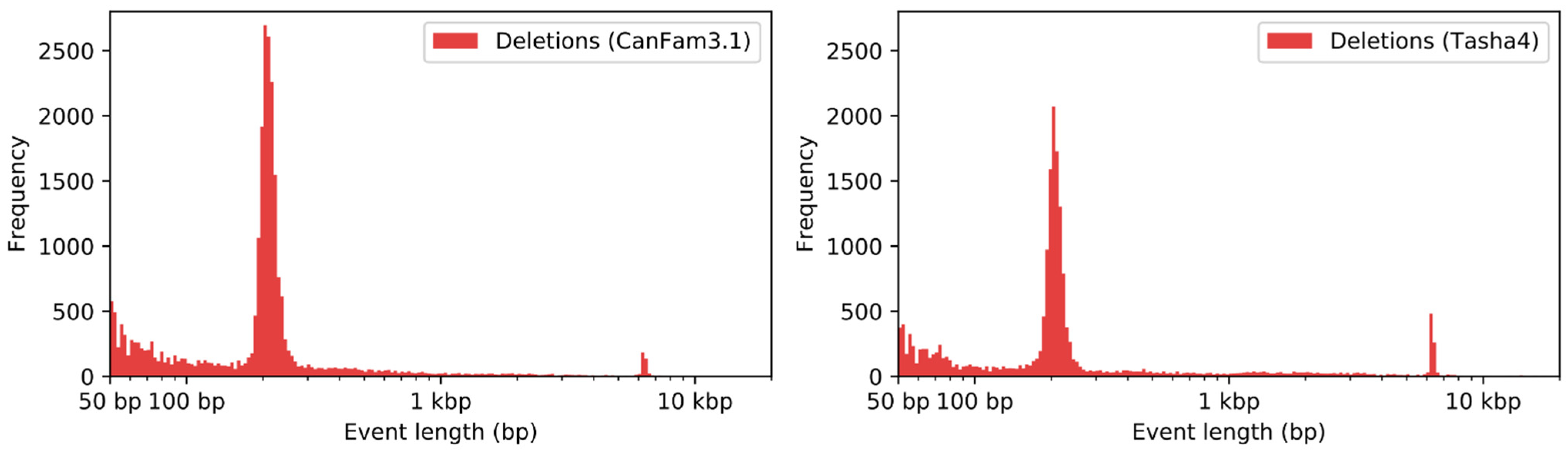
2.7. Structural Variant Detection
2.8. BAC Assembly
2.9. Mapping SNV Array Probes
3. Results
3.1. Dog10K_Boxer_Tasha_1.0 Assembly
3.2. Assembly Quality Assessment
3.3. Assembly Completeness
3.4. Gene Annotation
3.5. SNV Array Probes Mapped to Dog10k_Boxer_Tasha_1.0
3.6. Analysis of Duplications
3.7. Analysis of Repetitive Sequences
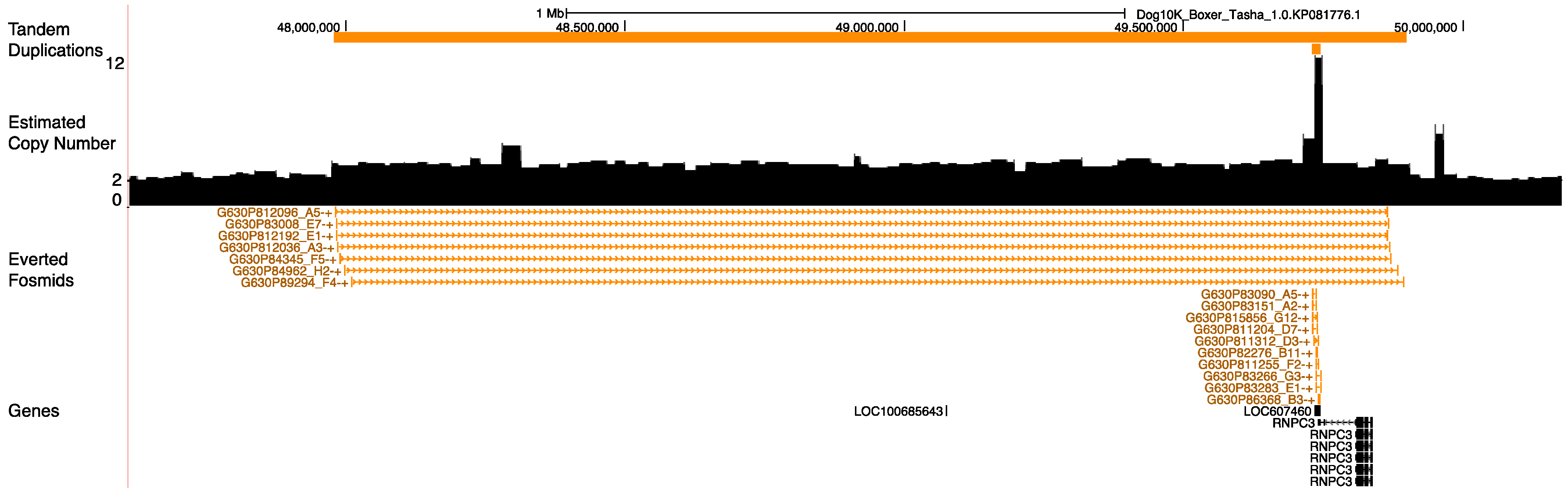
3.8. Duplications at the Pancreatic Amylase Locus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Xie, X.; Lu, J.; Kulbokas, E.J.; Golub, T.R.; Mootha, V.; Lindblad-Toh, K.; Lander, E.S.; Kellis, M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 2005, 434, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Dermitzakis, E.T.; Kirkness, E.; Schwarz, S.; Birney, E.; Reymond, A.; Antonarakis, S.E. Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 2004, 14, 852–859. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, O.; Olalde, I.; Berglund, J.; Lorente-Galdos, B.; Hernandez-Rodriguez, J.; Quilez, J.; Webster, M.T.; Wayne, R.K.; Lalueza-Fox, C.; Vilà, C.; et al. Analysis of structural diversity in wolf-like canids reveals post-domestication variants. BMC Genom. 2014, 15, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serres-Armero, A.; Povolotskaya, I.S.; Quilez, J.; Ramirez, O.; Santpere, G.; Kuderna, L.F.K.; Hernandez-Rodriguez, J.; Fernandez-Callejo, M.; Gomez-Sanchez, D.; Freedman, A.H.; et al. Similar genomic proportions of copy number variation within gray wolves and modern dog breeds inferred from whole genome sequencing. BMC Genom. 2017, 18, 977. [Google Scholar] [CrossRef] [Green Version]
- Halo, J.V.; Pendleton, A.L.; Shen, F.; Doucet, A.J.; Derrien, T.; Hitte, C.; Kirby, L.E.; Myers, B.; Sliwerska, E.; Emery, S.; et al. Long-read assembly of a great dane genome highlights the contribution of GC-rich sequence and mobile elements to canine genomes. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Field, M.A.; Ferguson, J.M.; Dudchenko, O.; Keilwagen, J.; Rosen, B.D.; Johnson, G.S.; Rice, E.S.; Hillier, L.D.; Hammond, J.M.; et al. Chromosome-length genome assembly and structural variations of the primal basenji dog (canis lupus familiaris) genome. BMC Genom. 2021, 22, 188. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wallerman, O.; Arendt, M.-L.; Sundström, E.; Karlsson, Å.; Nordin, J.; Mäkeläinen, S.; Pielberg, G.R.; Hanson, J.; Ohlsson, Å.; et al. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun. Biol. 2021, 4, 185. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with Wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Coombe, L.; Chu, J.; Warren, R.L.; Vandervalk, B.P.; Yeo, S.; Xue, Z.; Mohamadi, H.; Bohlmann, J.; Jones, S.J.M.; et al. Tigmint: Correcting assembly errors using linked reads from large molecules. BMC Bioinform. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Sahlin, K.; Chikhi, R.; Arvestad, L. Assembly scaffolding with PE-contaminated mate-pair libraries. Bioinformatics 2016, 32, 1925–1932. [Google Scholar] [CrossRef] [Green Version]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading Genomes with pacific biosciences RS long-read sequencing technology. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Hitte, C.; Madeoy, J.; Kirkness, E.F.; Priat, C.; Lorentzen, T.D.; Senger, F.; Thomas, D.; Derrien, T.; Ramirez, C.; Scott, C.; et al. Facilitating genome navigation: Survey sequencing and dense radiation-hybrid gene mapping. Nat. Rev. Genet. 2005, 6, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic. Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numanagic, I.; Gökkaya, A.S.; Zhang, L.; Berger, B.; Alkan, C.; Hach, F. Fast characterization of segmental duplications in genome assemblies. Bioinformatics 2018, 34, i706–i714. [Google Scholar] [CrossRef]
- Pendleton, A.L.; Shen, F.; Taravella, A.M.; Emery, S.; Veeramah, K.R.; Boyko, A.R.; Kidd, J.M. Comparison of village dog and wolf genomes highlights the role of the neural crest in dog domestication. BMC Biol. 2018, 16, 64. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Baertsch, R.; Hinrichs, A.; Miller, W.; Haussler, D. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 11484–11489. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI reference sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic. Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibaud-Nissen, F.; Souvorov, A.; Terence, M.; DiCuccio, M.; Kitts, P. The NCBI Handbook [Internet], 2nd ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2013. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK143764/ (accessed on 1 February 2021).
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT—The BLAST-like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.L.; Bhérer, C.; Morrow, B.E.; Boyko, A.R.; Auton, A. A Pedigree-based map of recombination in the domestic dog genome. G3 Genes Genomes Genet. 2016, 6, 3517–3524. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.K.; Ruhe, A.L.; Dumont, B.L.; Robertson, K.R.; Guerrero, G.; Shull, S.M.; Ziegle, J.S.; Millon, L.V.; Broman, K.W.; Payseur, B.A.; et al. A comprehensive linkage map of the dog genome. Genetics 2010, 184, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canis Lupus Familiaris Annotation Report. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genome/annotation_euk/Canis_lupus_familiaris/106/#TranscriptAlignmentStats (accessed on 15 April 2021).
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Perez, J.L.; Moran, J.V. The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Kirkness, E.F. Short Interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res. 2005, 15, 1798–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajic, P.; Pavlidis, P.; Dean, K.; Neznanova, L.; Romano, R.-A.; Garneau, D.; Daugherity, E.; Globig, A.; Ruhl, S.; Gokcumen, O. Independent amylase gene copy number bursts correlate with dietary preferences in mammals. Elife 2019, 8. [Google Scholar] [CrossRef]
- Axelsson, E.; Ratnakumar, A.; Arendt, M.-L.; Maqbool, K.; Webster, M.T.; Perloski, M.; Liberg, O.; Arnemo, J.M.; Hedhammar, A.; Lindblad-Toh, K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 2013, 495, 360–364. [Google Scholar] [CrossRef]
- Freedman, A.H.; Gronau, I.; Schweizer, R.M.; Vecchyo, D.O.-D.; Han, E.; Silva, P.M.; Galaverni, M.; Fan, Z.; Marx, P.; Lorente-Galdos, B.; et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014, 10, e1004016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollivier, M.; Tresset, A.; Bastian, F.; Lagoutte, L.; Axelsson, E.; Arendt, M.-L.; Bălăşescu, A.; Marshour, M.; Sablin, M.V.; Salanova, L.; et al. Amy2B copy number variation reveals starch diet adaptations in Ancient European dogs. R. Soc. Open Sci. 2016, 3, 160449. [Google Scholar] [CrossRef] [Green Version]
- Arendt, M.; Fall, T.; Lindblad-Toh, K.; Axelsson, E. Amylase activity is associated with AMY2B copy numbers in dog: Implications for dog domestication, diet and diabetes. Anim. Genet. 2014, 45, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Reiter, T.; Jagoda, E.; Capellini, T.D. Dietary variation and evolution of gene copy number among dog breeds. PLoS ONE 2016, 11, e0148899. [Google Scholar] [CrossRef] [PubMed]
- Field, M.A.; Rosen, B.D.; Dudchenko, O.; Chan, E.K.F.; Minoche, A.E.; Edwards, R.J.; Barton, K.; Lyons, R.J.; Tuipulotu, D.E.; Hayes, V.M.; et al. Canfam_GSD: De Novo chromosome-length genome assembly of the German Shepherd dog (Canis Lupus Familiaris) using a combination of long reads, optical mapping, and Hi-C. Gigascience 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Botigué, L.R.; Song, S.; Scheu, A.; Gopalan, S.; Pendleton, A.L.; Oetjens, M.; Taravella, A.M.; Seregély, T.; Zeeb-Lanz, A.; Arbogast, R.-M.; et al. Ancient european dog genomes reveal continuity since the Early Neolithic. Nat. Commun. 2017, 8, 16082. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Zerr, T.; Kidd, J.M.; Eichler, E.E.; Nickerson, D.A. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat. Genet. 2008, 40, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Grall, A.; Guaguère, E.; Planchais, S.; Grond, S.; Bourrat, E.; Hausser, I.; Hitte, C.; Le Gallo, M.; Derbois, C.; Kim, G.-J.; et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in Golden Retriever dogs and humans. Nat. Genet. 2012, 44, 140–147. [Google Scholar] [CrossRef]
- Zangerl, B.; Goldstein, O.; Philp, A.R.; Lindauer, S.J.P.; Pearce-Kelling, S.E.; Mullins, R.F.; Graphodatsky, A.S.; Ripoll, D.; Felix, J.S.; Stone, E.M.; et al. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics 2006, 88, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Kornegay, J.N. The Golden Retriever model of duchenne muscular dystrophy. Skelet. Muscle 2017, 7, 9. [Google Scholar] [CrossRef]
- Margolis, C.A.; Schneider, P.; Huttner, K.; Kirby, N.; Houser, T.P.; Wildman, L.; Grove, G.L.; Schneider, H.; Casal, M.L. Prenatal Treatment of X-linked hypohidrotic ectodermal dysplasia using recombinant ectodysplasin in a canine model. J. Pharmacol. Exp. Ther. 2019, 370, 806–813. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistic | CanFam3.1 | Dog10K_Boxer_Tasha_1.0 |
|---|---|---|
| Total sequence length | 2,410,976,875 | 2,312,802,206 |
| Total ungapped length | 2,392,715,236 | 2,312,743,367 |
| No. of scaffolds | 3310 | 147 |
| No. of unplaced scaffolds | 3228 | 107 |
| Scaffold N50 | 45,876,610 | 63,738,581 |
| Scaffold L50 | 20 | 14 |
| No. of unspanned gaps | 80 | 399 |
| No. of spanned gaps | 23,796 | 621 |
| No. of contigs | 27,106 | 1162 |
| Contig N50 | 267,478 | 27,487,084 |
| Contig L50 | 2436 | 31 |
| No. of chromosomes | 39 | 39 |
| Statistic | Dog10k_Boxer_Tasha_1.0 | CanFam3.1 |
|---|---|---|
| Complete BUSCOs | 95.3% | 92.2% |
| Complete and single copy BUSCOs | 94.1% | 91.1% |
| Complete and duplicated BUSCOs | 1.2% | 1.1% |
| Fragmented BUSCOs | 2.1% | 4.0% |
| Missing BUSCOs | 2.6% | 3.8% |
| Feature | Dog10k_Boxer_Tasha_1.0/Annotation Release 106 |
|---|---|
| Protein-coding genes | 20,100 |
| Non-coding genes | 15,306 |
| Small non-coding genes | 2083 |
| Long non-coding genes | 12,667 |
| Miscellaneous * non-coding genes | 10 |
| Pseudogenes | 4887 |
| Dog10K_Boxer_Tasha_1.0 | CanFam3.1 | |||
|---|---|---|---|---|
| Repeat Class | Elements | bp | Elements | bp |
| DNA | 341,866 | 65,043,282 | 347,025 | 65,997,048 |
| LINE | 1,286,663 | 467,394,285 | 1,307,498 | 470,518,469 |
| LTR | 378,505 | 111,520,139 | 384,551 | 113,151,392 |
| Low_complexity | 123,075 | 6,525,287 | 120,803 | 6,009,804 |
| RC | 1636 | 345,889 | 1649 | 347,342 |
| RNA | 489 | 103,097 | 504 | 105,770 |
| SINE | 1,579,792 | 240,791,186 | 1,605,511 | 244,461,861 |
| Satellite | 5730 | 11,298,647 | 635 | 624,881 |
| Simple_repeat | 891,331 | 40,450,974 | 895,091 | 38,358,719 |
| Unknown | 3449 | 559,562 | 3487 | 565,722 |
| rRNA | 953 | 129,078 | 958 | 115,711 |
| scRNA | 70 | 4996 | 71 | 5156 |
| snRNA | 4492 | 278,022 | 4617 | 285,578 |
| srpRNA | 45 | 8900 | 47 | 9496 |
| tRNA | 35,501 | 2,608,084 | 35,906 | 2,636,278 |
| Dog10K_Boxer_Tasha_1.0 | CanFam3.1 | |||
|---|---|---|---|---|
| Repeat Class | Elements | bp | Elements | bp |
| SINEC | 1,125,416 | 177,104,238 | 1,146,663 | 180,147,553 |
| SINEC < 10% divergence | 454,869 | 71,490,885 | 464,113 | 72,819,234 |
| LINE/L1 | 853,212 | 379,452,954 | 869,259 | 381,738,114 |
| LINE/L1 < 10% divergence and ≥4 kb | 4805 | 26,935,018 | 4229 | 23,359,516 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagannathan, V.; Hitte, C.; Kidd, J.M.; Masterson, P.; Murphy, T.D.; Emery, S.; Davis, B.; Buckley, R.M.; Liu, Y.-H.; Zhang, X.-Q.; et al. Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome. Genes 2021, 12, 847. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060847
Jagannathan V, Hitte C, Kidd JM, Masterson P, Murphy TD, Emery S, Davis B, Buckley RM, Liu Y-H, Zhang X-Q, et al. Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome. Genes. 2021; 12(6):847. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060847
Chicago/Turabian StyleJagannathan, Vidhya, Christophe Hitte, Jeffrey M. Kidd, Patrick Masterson, Terence D. Murphy, Sarah Emery, Brian Davis, Reuben M. Buckley, Yan-Hu Liu, Xiang-Quan Zhang, and et al. 2021. "Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome" Genes 12, no. 6: 847. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060847