
Analysis of Worldwide Carrier Frequency and Predicted Genetic Prevalence of Autosomal Recessive Congenital Hypothyroidism Based on a General Population Database


Abstract
:1. Introduction
2. Materials and Methods
2.1. Database Analysis
2.2. Genetic Variant Classification
2.3. Carrier Frequency (CF) and Predicted Genetic Prevalence Analysis (pGP)
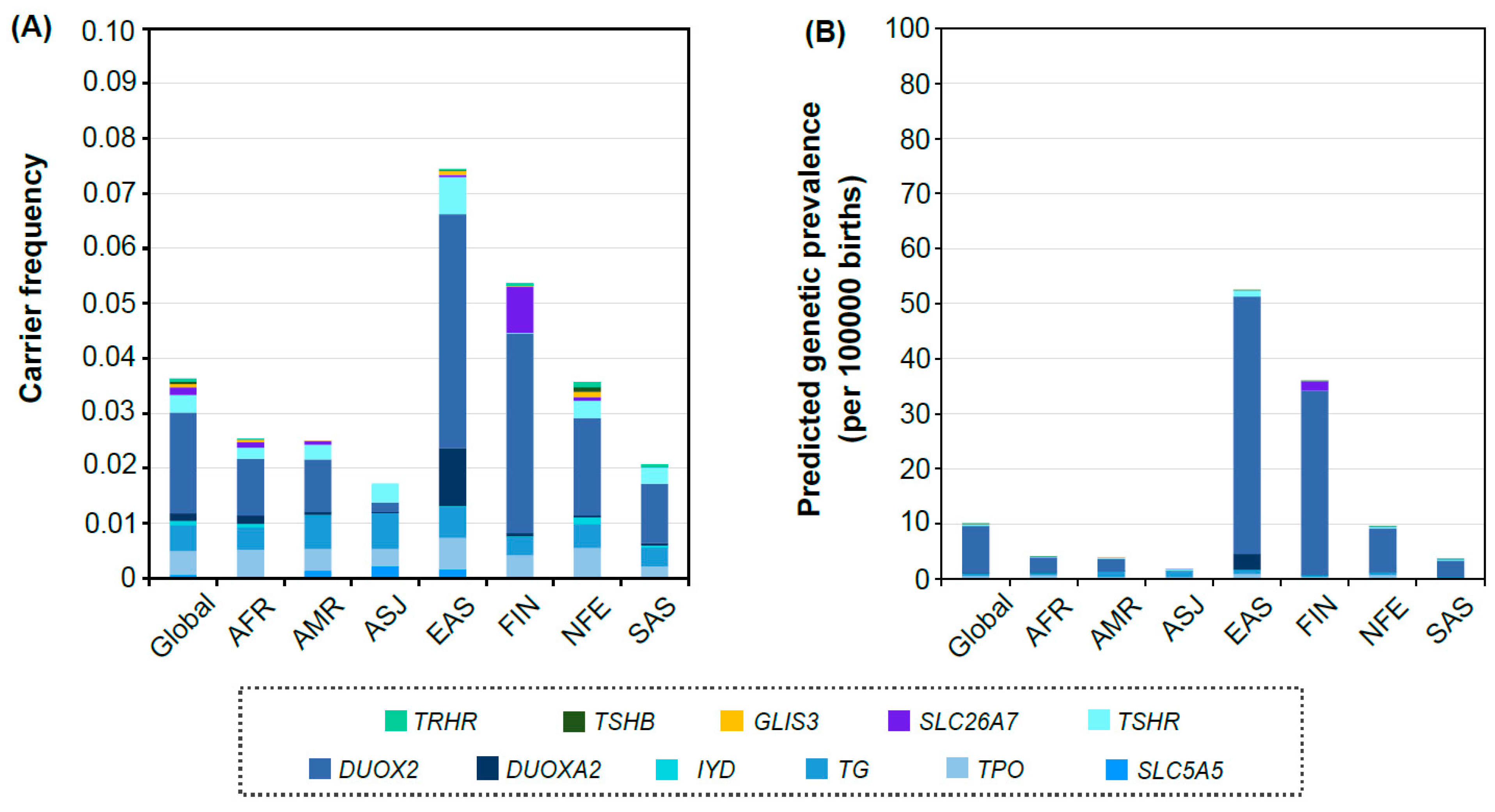
3. Results
3.1. Presumed (Likely) Pathogenic Variants in 12 Candidate Genes
3.2. Distribution of Carrier Frequency and Predicted Genetic Prevalence in Each Population Group
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andermann, A.; Blancquaert, I. Genetic screening: A primer for primary care. Can. Fam. Physician 2010, 56, 333–339. [Google Scholar] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Büyükgebiz, A. Newborn Screening for Congenital Hypothyroidism. J. Clin. Res. Pediatr. Endocrinol. 2013, 5 (Suppl. 1), 8–12. [Google Scholar] [CrossRef] [PubMed]
- Dunnen, J.T.D.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.; et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Tayoun, A.N.A.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Zastrow, D.B.; Baudet, H.; Shen, W.; Thomas, A.; Si, Y.; Weaver, M.A.; Lager, A.M.; Liu, J.; Mangels, R.; Dwight, S.S.; et al. Unique aspects of sequence variant interpretation for inborn errors of metabolism (IEM): The ClinGen IEM Working Group and the Phenylalanine Hydroxylase Gene. Hum. Mutat. 2018, 39, 1569–1580. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanany, M.; Allon, G.; Kimchi, A.; Blumenfeld, A.; Newman, H.; Pras, E.; Wormser, O.; Birk, O.S.; Gradstein, L.; Banin, E.; et al. Carrier frequency analysis of mutations causing autosomal-recessive-inherited retinal diseases in the Israeli population. Eur. J. Hum. Genet. 2018, 26, 1159–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Park, K.S. Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Mol. Genet. Metab. Rep. 2021, 27, 100734. [Google Scholar] [CrossRef]
- Feuchtbaum, L.; Carter, J.; Dowray, S.; Currier, R.J.; Lorey, F. Birth prevalence of disorders detectable through newborn screening by race/ethnicity. Genet. Med. 2012, 14, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Cherella, C.E.; Wassner, A.J. Congenital hypothyroidism: Insights into pathogenesis and treatment. Int. J. Pediatr. Endocrinol. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, D.; Polizzotti, N.; Carta, A.; Gelsomino, R.; Sava, L.; Vigneri, R.; Calaciura, F. Longitudinal Study of Thyroid Function in Children with Mild Hyperthyrotropinemia at Neonatal Screening for Congenital Hypothyroidism. J. Clin. Endocrinol. Metab. 2008, 93, 2679–2685. [Google Scholar] [CrossRef]
- Lain, S.J.; Bentley, J.P.; Wiley, V.; Roberts, C.L.; Jack, M.; Wilcken, B.; Nassar, N. Association between borderline neonatal thyroid-stimulating hormone concentrations and educational and developmental outcomes: A population-based record-linkage study. Lancet Diabetes Endocrinol. 2016, 4, 756–765. [Google Scholar] [CrossRef]
- Wassner, A.J.; Brown, R.S. Congenital hypothyroidism: Recent advances. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 407–412. [Google Scholar] [CrossRef]
- Olivieri, A.; Fazzini, C.; Medda, E. The Italian Study Group for Congenital Hypothyroidism Multiple Factors Influencing the Incidence of Congenital Hypothyroidism Detected by Neonatal Screening. Horm. Res. Paediatr. 2015, 83, 86–93. [Google Scholar] [CrossRef]
- Deladoëy, J.; Ruel, J.; Giguère, Y.; van Vliet, G. Is the Incidence of Congenital Hypothyroidism Really Increasing? A 20-Year Retrospective Population-Based Study in Québec. J. Clin. Endocrinol. Metab. 2011, 96, 2422–2429. [Google Scholar] [CrossRef] [Green Version]
- Van Trotsenburg, P.; Stoupa, A.; Léger, J.; Rohrer, T.; Peters, C.; Fugazzola, L.; Cassio, A.; Heinrichs, C.; Beauloye, V.; Pohlenz, J.; et al. Congenital Hypothyroidism: A 2020–2021 Consensus Guidelines Update—An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid 2021, 31, 387–419. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; van Trotsenburg, A.S.P.; Schoenmakers, N. Diagnosis of Endocrine Disease: Congenital hypothyroidism: Update and perspectives. Eur. J. Endocrinol. 2018, 179, R297–R317. [Google Scholar] [CrossRef] [Green Version]
- Stoppa-Vaucher, S.; van Vliet, G.; Deladoëy, J. Variation by Ethnicity in the Prevalence of Congenital Hypothyroidism Due to Thyroid Dysgenesis. Thyroid 2011, 21, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, C.F.; Harris, K.B.; Borgfeld, L.; Drummond-Borg, M.; Eaton, R.; Lorey, F.; Therrell, B.L.; Wallace, J.; Pass, K.A. Trends in Incidence Rates of Congenital Hypothyroidism Related to Select Demographic Factors: Data from the United States, California, Massachusetts, New York, and Texas. Pediatrics 2010, 125 (Suppl. 2), S37–S47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Zhang, J.-X.; Yang, C.-Y.; Gao, G.-Q.; Zhu, W.-B.; Han, B.; Zhang, L.-L.; Wan, Y.-Y.; Ye, X.-P.; Ma, Y.-R.; et al. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur. J. Endocrinol. 2018, 178, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.-J.; Park, H.-K.; Kim, Y.-J.; Lee, K.-R.; Park, J.-H.; Park, J.-H.; Park, H.-D.; Lee, S.-Y.; Kim, J.-W. DUOX2 Mutations Are Frequently Associated with Congenital Hypothyroidism in the Korean Population. Ann. Lab. Med. 2016, 36, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Long, W.; Yang, Y.; Wang, Y.; Jiang, L.; Cai, Z.; Wang, H. Newborn Screening and Molecular Profile of Congenital Hypothyroidism in a Chinese Population. Front. Genet. 2018, 9, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cangul, H.; Liao, X.-H.; Schoenmakers, E.; Kero, J.; Barone, S.; Srichomkwun, P.; Iwayama, H.; Serra, E.G.; Saglam, H.; Eren, E.; et al. Homozygous loss-of-function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoupa, A.; Chehade, G.A.H.; Chaabane, R.; Kariyawasam, D.; Szinnai, G.; Hanein, S.; Bole-Feysot, C.; Fourrage, C.; Nitschke, P.; Thalassinos, C.; et al. High Diagnostic Yield of Targeted Next-Generation Sequencing in a Cohort of Patients with Congenital Hypothyroidism Due to Dyshormonogenesis. Front. Endocrinol. 2020, 11, 545339. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, H.Y.; Kim, Y.M.; Lee, H.; Bae, M.H.; Park, K.H.; Lee, S.-M.; Kwak, M.J. Genetic Evaluation of Congenital Hypothyroidism with Gland in situ Using Targeted Exome Sequencing. Ann. Clin. Lab. Sci. 2021, 51, 73–81. [Google Scholar]
- Makretskaya, N.; Bezlepkina, O.; Kolodkina, A.; Kiyaev, A.; Vasilyev, E.V.; Petrov, V.; Kalinenkova, S.; Malievsky, O.; Dedov, I.I.; Tiulpakov, A. High frequency of mutations in ’dyshormonogenesis genes’ in severe congenital hypothyroidism. PLoS ONE 2018, 13, e0204323. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Lu, G.; Zhou, W.; Yang, Y.; Zhang, B.; Zhou, H.; Jiang, L.; Yu, B. Targeted next-generation sequencing of thirteen causative genes in Chinese patients with congenital hypothyroidism. Endocr. J. 2018, 65, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Aycan, Z.; Cangul, H.; Muzza, M.; Bas, V.N.; Fugazzola, L.; Chatterjee, V.K.; Persani, L.; Schoenmakers, N. Digenic DUOX1 and DUOX2 Mutations in Cases with Congenital Hypothyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 3085–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medda, E.; Olivieri, A.; Stazi, M.A.; Grandolfo, M.E.; Fazzini, C.; Baserga, M.; Burroni, M.; Cacciari, E.; Calaciura, F.; Cassio, A.; et al. Risk factors for congenital hypothyroidism: Results of a population case-control study (1997–2003). Eur. J. Endocrinol. 2005, 153, 765–773. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Group | Gene | No. of Presumed Pathogenic or Likely Pathogenic Variants | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Global | AFR | AMR | ASJ | EAS | FIN | NFE | SAS | ||
| DH | SLC5A5 | 18 | 1 | 3 | 2 | 5 | 8 | 6 | 3 |
| TPO | 79 | 15 | 13 | 4 | 9 | 3 | 51 | 9 | |
| TG | 163 | 25 | 29 | 5 | 25 | 5 | 79 | 32 | |
| IYD | 20 | 5 | 1 | 0 | 1 | 1 | 14 | 3 | |
| DUOXA2 | 34 | 8 | 4 | 1 | 4 | 1 | 12 | 4 | |
| DUOX2 | 143 | 28 | 37 | 3 | 39 | 6 | 74 | 24 | |
| TSHR | 53 | 14 | 11 | 1 | 5 | 2 | 33 | 6 | |
| SLC26A7 | 46 | 8 | 8 | 1 | 6 | 1 | 27 | 5 | |
| TD | GLIS3 | 28 | 5 | 3 | 0 | 4 | 3 | 18 | 0 |
| FOXE1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ICCH | TSHB | 8 | 1 | 0 | 0 | 1 | 1 | 6 | 1 |
| TRHR | 18 | 1 | 0 | 0 | 1 | 4 | 15 | 1 | |
| Genes 1 | Variant | Allele frequency in gnomAD (v2.1.1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Major Population 2 | Global | AFR | AMR | ASJ | EAS | FIN | NFE | SAS | ||
| TPO | c.483-1G>C | AFR | 0.00013 | 0.00133 | 0.00004 | 0 | 0 | 0 | 0.00001 | 0 |
| c.1184_1187dupGCCG, (p.Ala397ProfsTer76) | FIN, NFE, AMR | 0.00053 | 0 | 0.00037 | 0 | 0 | 0.00156 | 0.00092 | 0 | |
| c.1978C>G, (p.Gln660Glu) | AMR, NFE | 0.00030 | 0.00008 | 0.00119 | 0 | 0 | 0 | 0.00023 | 0 | |
| c.2268dupT, (p.Glu757Ter) | EAS | 0.00012 | 0 | 0 | 0 | 0.00165 | 0 | 0 | 0 | |
| TG | c.886C>T, (p.Arg296Ter) | ASJ, AMR, NFE, AFR | 0.00035 | 0.00028 | 0.00059 | 0.00106 | 0 | 0.00004 | 0.00042 | 0.00003 |
| c.1963C>T, (p.Gln655Ter) | FIN | 0.00013 | 0 | 0 | 0 | 0 | 0.00140 | 0.00002 | 0 | |
| c.5184C>A, (p.Cys1728Ter) | ASJ | 0.00010 | 0 | 0.00003 | 0.00188 | 0 | 0 | 0.00003 | 0 | |
| IYD | c.315_317delCAT, (p.Phe105_Ile106delinsLeu) | NFE | 0.00011 | 0.00008 | 0 | 0 | 0 | 0 | 0.00022 | 0 |
| DUOXA2 | c.413dupA, (p.Tyr138Ter) | EAS | 0.00023 | 0 | 0 | 0 | 0.00333 | 0 | 0 | 0 |
| c.738C>G, (p.Tyr246Ter) | EAS | 0.00014 | 0 | 0 | 0 | 0.00186 | 0 | 0.00001 | 0 | |
| DUOX2 | c.4524+1dupG | AFR | 0.00011 | 0.00126 | 0.00003 | 0 | 0 | 0 | 0 | 0 |
| c.3693+1G>T | EAS | 0.00011 | 0 | 0 | 0 | 0.00160 | 0 | 0 | 0 | |
| c.3329G>A, (p.Arg1110Gln) | EAS | 0.00019 | 0 | 0.00003 | 0 | 0.00251 | 0 | 0.00002 | 0 | |
| c.3155G>A, (p.Cys1052Tyr) | FIN, NFE, AMR, AFR | 0.00128 | 0.00012 | 0.00014 | 0.00010 | 0 | 0.00629 | 0.00140 | 0 | |
| c.2895_2898delGTTC, (p.Phe966SerfsTer29) | FIN, NFE, SAS, AMR, AFR | 0.00290 | 0.00056 | 0.00158 | 0 | 0 | 0.01139 | 0.00290 | 0.00213 | |
| c.1588A>T, (p.Lys530Ter) | EAS | 0.00063 | 0 | 0 | 0 | 0.00882 | 0 | 0 | 0 | |
| c.1516G>A, (p.Asp506Asn) | NFE, FIN | 0.00026 | 0.00004 | 0.00003 | 0 | 0 | 0.00040 | 0.00047 | 0 | |
| c.1462G>A, (p.Gly488Arg) | EAS, AFR | 0.00015 | 0.00052 | 0 | 0 | 0.00120 | 0 | 0.00004 | 0 | |
| c.1060C>T, (p.Arg354Trp) | AMR, SAS, NFE | 0.00016 | 0.00008 | 0.00037 | 0 | 0.00005 | 0 | 0.00012 | 0.00029 | |
| c.602dupG, (p.Gln202ThrfsTer99) | NFE, SAS, AMR, AFR, FIN | 0.00086 | 0.00032 | 0.00055 | 0 | 0 | 0.00012 | 0.00158 | 0.00070 | |
| TSHR | c.202C>T, (p.Pro68Ser) | ASJ, SAS, AMR, NFE | 0.00049 | 0.00004 | 0.00068 | 0.00174 | 0 | 0 | 0.00043 | 0.00115 |
| c.484C>G, (p.Pro162Ala) | NFE, EAS | 0.00014 | 0.00004 | 0.00008 | 0 | 0.00010 | 0 | 0.00025 | 0 | |
| c.1349G>A, (p.Arg450His) | EAS, SAS | 0.00021 | 0 | 0.00008 | 0 | 0.00241 | 0.00004 | 0.00003 | 0.00013 | |
| c.1637G>A, (p.Trp546Ter) | NFE | 0.00011 | 0.00008 | 0 | 0 | 0 | 0 | 0.00022 | 0 | |
| SLC26A7 | c.1893delT, (p.Phe631LeufsTer8) | FIN, NFE | 0.00045 | 0.00004 | 0.00003 | 0 | 0 | 0.00420 | 0.00010 | 0 |
| TSHB | c.373delT, (p.Cys125ValfsTer10) | NFE | 0.00016 | 0.00004 | 0 | 0 | 0 | 0.00004 | 0.00032 | 0 |
| TRHR | c.1016delA, (p.Gln339ArgfsTer14) | NFE | 0.00012 | 0.00004 | 0 | 0 | 0 | 0 | 0.00024 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.-S. Analysis of Worldwide Carrier Frequency and Predicted Genetic Prevalence of Autosomal Recessive Congenital Hypothyroidism Based on a General Population Database. Genes 2021, 12, 863. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060863
Park K-S. Analysis of Worldwide Carrier Frequency and Predicted Genetic Prevalence of Autosomal Recessive Congenital Hypothyroidism Based on a General Population Database. Genes. 2021; 12(6):863. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060863
Chicago/Turabian StylePark, Kyung-Sun. 2021. "Analysis of Worldwide Carrier Frequency and Predicted Genetic Prevalence of Autosomal Recessive Congenital Hypothyroidism Based on a General Population Database" Genes 12, no. 6: 863. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060863