
The Combined Human Genotype of Truncating TTN and RBM20 Mutations Is Associated with Severe and Early Onset of Dilated Cardiomyopathy

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Description of the Patients
2.2. Genetic Analyses
2.3. Preparation of Myocardial Tissue
2.4. Isolation of Total RNA
2.5. Quantitative Real Time Polymerase Chain Reaction
2.6. Plasmid Construction
2.7. Cell Culture and Transient Transfection
2.8. Immunohistochemistry
2.9. Confocal Microscopy
2.10. RNA-Sequencing
3. Results
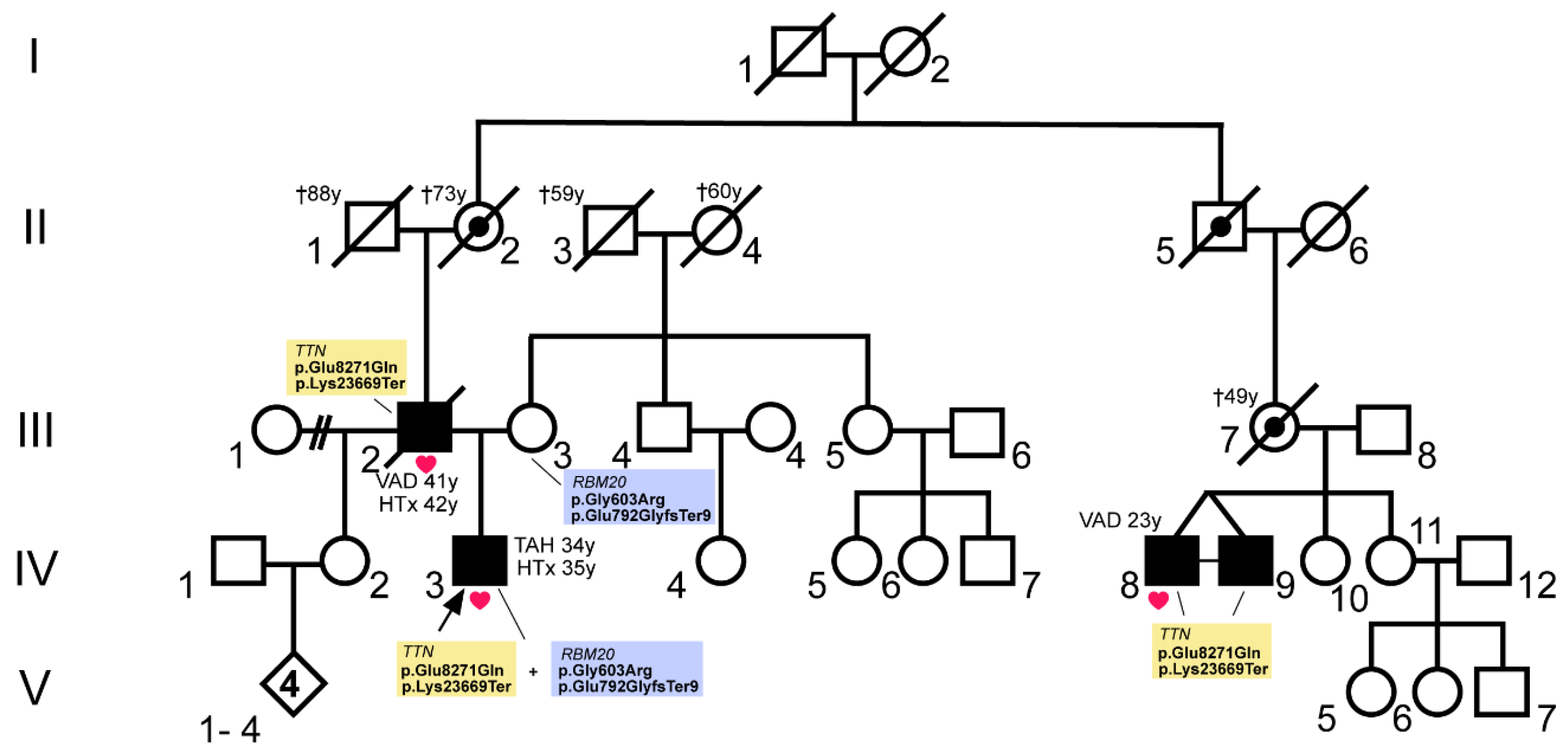
3.1. A Combination of Truncating RBM20 and TTN Variants Was Identified in a Patient with Severe DCM
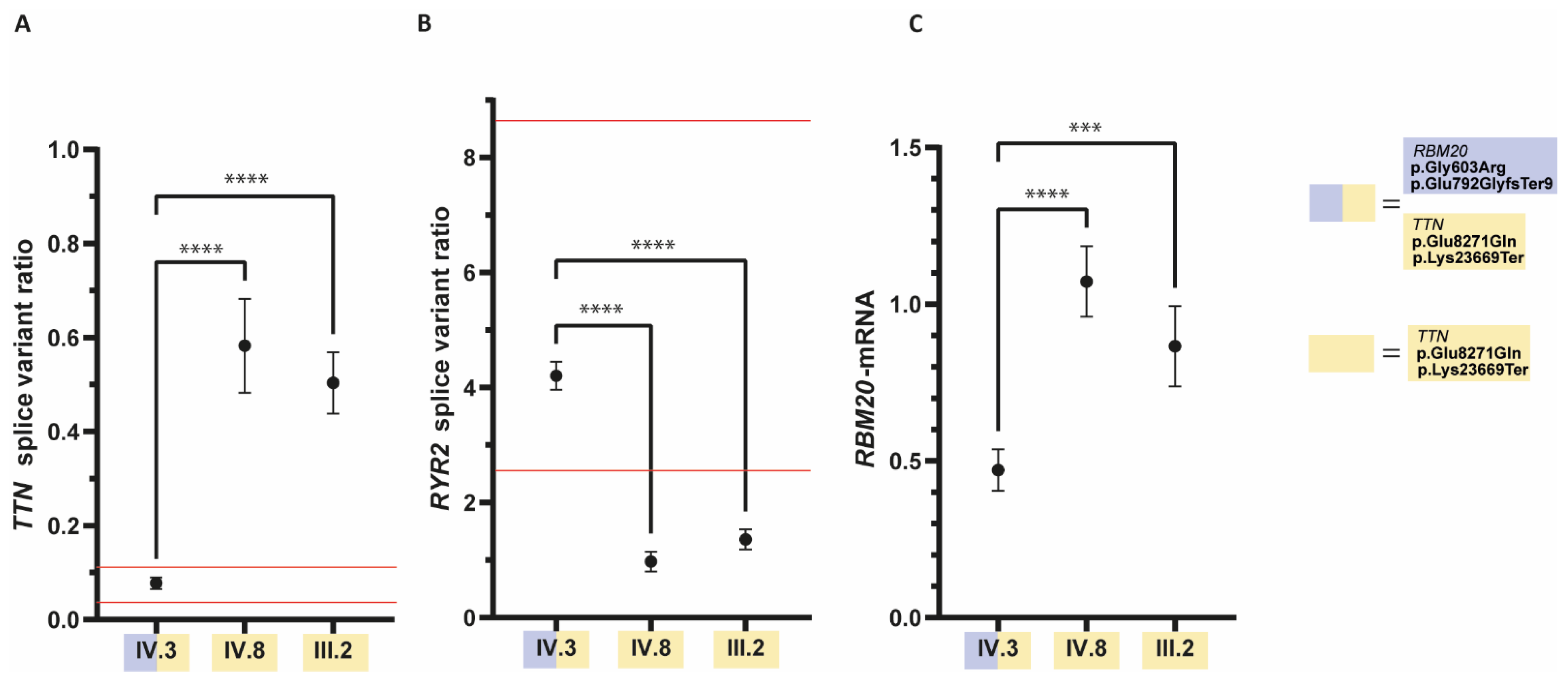
3.2. The Ratio of RYR2- and TTN-Splice Variants Is Altered in the Patient with the RBM20 Mutations
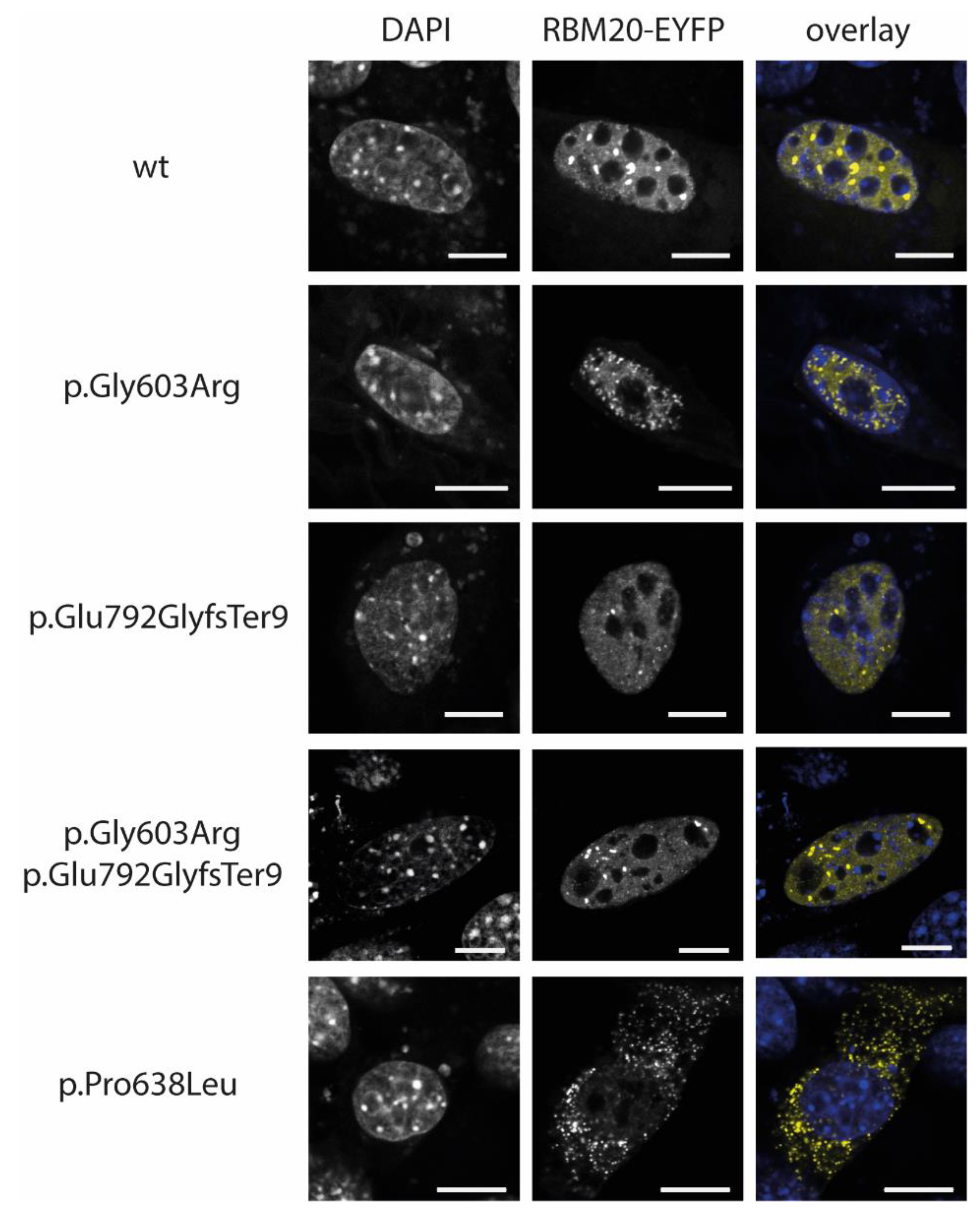
3.3. RBM20-p.Gly603Arg and -p.Glu792GlyfsTer9 Do Not Lead to an Abnormal Cytoplasmic Mislocalization
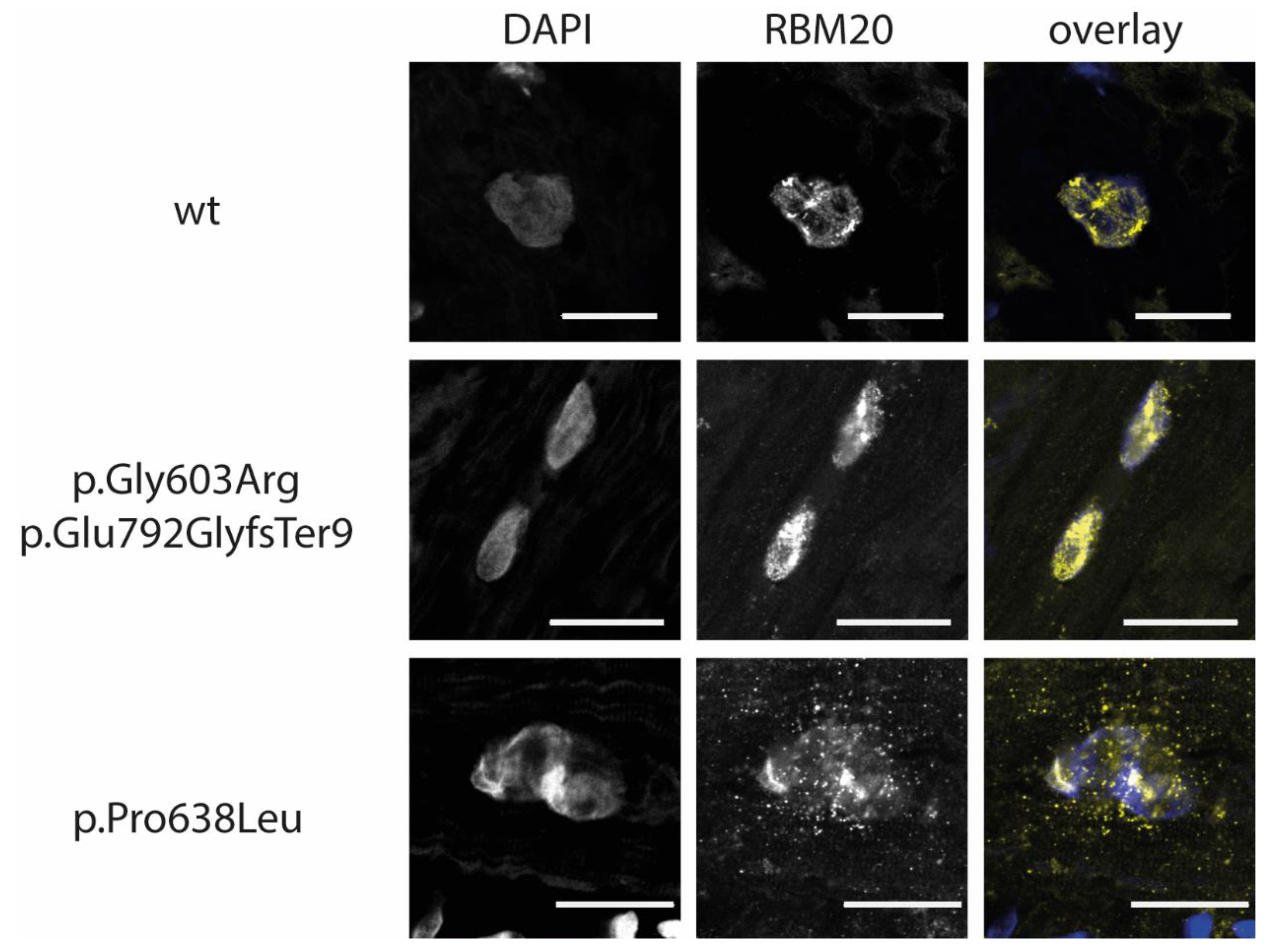
3.4. RBM20-p.Gly603Arg and -p.Glu792GlyfsTer9 Lead to RBM20 Haploinsufficiency in the Index Patient
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Khush, K.K.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Hayes, D., Jr.; Kucheryavaya, A.Y.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Stehlik, J.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-fifth Adult Heart Transplantation Report-2018; Focus Theme: Multiorgan Transplantation. J. Heart Lung Transpl. 2018, 37, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Rossano, J.W.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Hayes, D., Jr.; Khush, K.K.; Kucheryavaya, A.Y.; Toll, A.E.; Levvey, B.J.; Meiser, B.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Twenty-first pediatric heart transplantation report-2018; Focus theme: Multiorgan Transplantation. J. Heart Lung Transpl. 2018, 37, 1184–1195. [Google Scholar] [CrossRef]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Bohm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ebbinghaus, H.; Deutsch, M.A.; Gummert, J.; Gartner, A.; Ratnavadivel, S.; Milting, H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381. [Google Scholar] [CrossRef] [Green Version]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. RBM20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [Green Version]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.O.; et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum. Mutat. 2020, 41, 1931–1943. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Filippello, A.; Lorenzi, P.; Bergamo, E.; Romanelli, M.G. Identification of nuclear retention domains in the RBM20 protein. FEBS Lett. 2013, 587, 2989–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeland, C.J.; van den Hoogenhof, M.M.; Beqqali, A.; Creemers, E.E. Insights into alternative splicing of sarcomeric genes in the heart. J. Mol. Cell. Cardiol. 2015, 81, 107–113. [Google Scholar] [CrossRef]
- Beqqali, A.; Bollen, I.A.; Rasmussen, T.B.; van den Hoogenhof, M.M.; van Deutekom, H.W.; Schafer, S.; Haas, J.; Meder, B.; Sorensen, K.E.; van Oort, R.J.; et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 2016, 112, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Marschall, C.; Moscu-Gregor, A.; Klein, H.G. Variant panorama in 1385 index patients and sensitivity of expanded next-generation sequencing panels in arrhythmogenic disorders. Cardiovasc. Diagn. Ther. 2019, 9, S292–S298. [Google Scholar] [CrossRef] [PubMed]
- Waldmuller, S.; Schroeder, C.; Sturm, M.; Scheffold, T.; Imbrich, K.; Junker, S.; Frische, C.; Hofbeck, M.; Bauer, P.; Bonin, M.; et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol. Cell. Probes 2015, 29, 308–314. [Google Scholar] [CrossRef]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012, 9, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Xie, Y.; Zhang, H.; Feng, Z.; Huang, J.; Huang, J.; Hu, S.; Wei, Y. Multiple genetic variants in adolescent patients with left ventricular noncompaction cardiomyopathy. Int. J. Cardiol. 2020, 302, 117–123. [Google Scholar] [CrossRef]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.; et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Jorda, P.; Toro, R.; Diez, C.; Salazar-Mendiguchia, J.; Fernandez-Falgueras, A.; Perez-Serra, A.; Coll, M.; Puigmule, M.; Arbelo, E.; Garcia-Alvarez, A.; et al. Malignant Arrhythmogenic Role Associated with RBM20: A Comprehensive Interpretation Focused on a Personalized Approach. J. Pers. Med. 2021, 11, 130. [Google Scholar] [CrossRef]
- Gaertner, A.; Klauke, B.; Brodehl, A.; Milting, H. RBM20 mutations in left ventricular non-compaction cardiomyopathy. Pediatr. Investig. 2020, 4, 61–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.R.; Granzier, H.L. Titin-based tension in the cardiac sarcomere: Molecular origin and physiological adaptations. Prog. Biophys. Mol. Biol. 2012, 110, 204–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, W.A. Titin Gene and Protein Functions in Passive and Active Muscle. Annu. Rev. Physiol. 2018, 80, 389–411. [Google Scholar] [CrossRef]
- Siu, B.L.; Niimura, H.; Osborne, J.A.; Fatkin, D.; MacRae, C.; Solomon, S.; Benson, D.W.; Seidman, J.G.; Seidman, C.E. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation 1999, 99, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Atherton, J.; Geupel, A.; Sasse-Klaassen, S.; Heuser, A.; Frenneaux, M.; McNabb, M.; Granzier, H.; Labeit, S.; Thierfelder, L. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J. Mol. Med. 2006, 84, 478–483. [Google Scholar] [CrossRef]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra276. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.S.; Seidman, J.G.; Arany, Z. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 2601–2602. [Google Scholar] [CrossRef]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, A.; Wilke, A.; Komajda, M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 1999, 20, 93–102. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- World Medical, A. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [Green Version]
- Gaertner-Rommel, A.; Tiesmeier, J.; Jakob, T.; Strickmann, B.; Veit, G.; Bachmann-Mennenga, B.; Paluszkiewicz, L.; Klingel, K.; Schulz, U.; Laser, K.T.; et al. Molecular autopsy and family screening in a young case of sudden cardiac death reveals an unusually severe case of FHL1 related hypertrophic cardiomyopathy. Mol. Genet. Genom. Med. 2019, 7, e841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sielemann, K.; Elbeck, Z.; Gartner, A.; Brodehl, A.; Stanasiuk, C.; Fox, H.; Paluszkiewicz, L.; Tiesmeier, J.; Wlost, S.; Gummert, J.; et al. Distinct Myocardial Transcriptomic Profiles of Cardiomyopathies Stratified by the Mutant Genes. Genes 2020, 11, 1430. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Murayama, R.; Kimura-Asami, M.; Togo-Ohno, M.; Yamasaki-Kato, Y.; Naruse, T.K.; Yamamoto, T.; Hayashi, T.; Ai, T.; Spoonamore, K.G.; Kovacs, R.J.; et al. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci. Rep. 2018, 8, 8970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Ebbinghaus, H.; Gaertner-Rommel, A.; Stanasiuk, C.; Klauke, B.; Milting, H. Functional analysis of DES-p.L398P and RBM20-p.R636C. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.W.; Oommen, S.; Qureshi, M.Y.; Goetsch, S.C.; Pease, D.R.; Sundsbak, R.S.; Guo, W.; Sun, M.; Sun, H.; Kuroyanagi, H.; et al. Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat. Med. 2020, 26, 1788–1800. [Google Scholar] [CrossRef]
- Ihara, K.; Sasano, T.; Hiraoka, Y.; Togo-Ohno, M.; Soejima, Y.; Sawabe, M.; Tsuchiya, M.; Ogawa, H.; Furukawa, T.; Kuroyanagi, H. A missense mutation in the RSRSP stretch of RBM20 causes dilated cardiomyopathy and atrial fibrillation in mice. Sci. Rep. 2020, 10, 17894. [Google Scholar] [CrossRef]
- Frischmeyer, P.A.; Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Maquat, L.E. Evidence that the decay of nucleus-associated nonsense mRNA for human triosephosphate isomerase involves nonsense codon recognition after splicing. RNA 1996, 2, 235–243. [Google Scholar] [PubMed]
- Popp, M.W.; Maquat, L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013, 47, 139–165. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Thermann, R.; Neu-Yilik, G.; Deters, A.; Frede, U.; Wehr, K.; Hagemeier, C.; Hentze, M.W.; Kulozik, A.E. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998, 17, 3484–3494. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working, G. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Muller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Akinrinade, O.; Ollila, L.; Vattulainen, S.; Tallila, J.; Gentile, M.; Salmenpera, P.; Koillinen, H.; Kaartinen, M.; Nieminen, M.S.; Myllykangas, S.; et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur. Heart J. 2015, 36, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; Restrepo-Cordoba, M.A.; Dal Ferro, M.; Stolfo, D.; Johnson, R.; et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ. Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [Green Version]
- van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knoll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef] [Green Version]
- Won, S.; Hong, R.A.; Shohet, R.V.; Seto, T.B.; Parikh, N.I. Methamphetamine-associated cardiomyopathy. Clin. Cardiol. 2013, 36, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.J. Reversible dilated cardiomyopathy induced by methamphetamine. Clin. Cardiol. 1989, 12, 725–727. [Google Scholar] [CrossRef]
- Ito, H.; Yeo, K.K.; Wijetunga, M.; Seto, T.B.; Tay, K.; Schatz, I.J. A comparison of echocardiographic findings in young adults with cardiomyopathy: With and without a history of methamphetamine abuse. Clin. Cardiol. 2009, 32, E18–E22. [Google Scholar] [CrossRef]
- Yeo, K.K.; Wijetunga, M.; Ito, H.; Efird, J.T.; Tay, K.; Seto, T.B.; Alimineti, K.; Kimata, C.; Schatz, I.J. The association of methamphetamine use and cardiomyopathy in young patients. Am. J. Med. 2007, 120, 165–171. [Google Scholar] [CrossRef]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Molgaard, H.; Moller, J.E.; Eiskjaer, H.; Mogensen, J. Pathogenic RBM20-Variants Are Associated With a Severe Disease Expression in Male Patients With Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaertner, A.; Bloebaum, J.; Brodehl, A.; Klauke, B.; Sielemann, K.; Kassner, A.; Fox, H.; Morshuis, M.; Tiesmeier, J.; Schulz, U.; et al. The Combined Human Genotype of Truncating TTN and RBM20 Mutations Is Associated with Severe and Early Onset of Dilated Cardiomyopathy. Genes 2021, 12, 883. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060883
Gaertner A, Bloebaum J, Brodehl A, Klauke B, Sielemann K, Kassner A, Fox H, Morshuis M, Tiesmeier J, Schulz U, et al. The Combined Human Genotype of Truncating TTN and RBM20 Mutations Is Associated with Severe and Early Onset of Dilated Cardiomyopathy. Genes. 2021; 12(6):883. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060883
Chicago/Turabian StyleGaertner, Anna, Julia Bloebaum, Andreas Brodehl, Baerbel Klauke, Katharina Sielemann, Astrid Kassner, Henrik Fox, Michiel Morshuis, Jens Tiesmeier, Uwe Schulz, and et al. 2021. "The Combined Human Genotype of Truncating TTN and RBM20 Mutations Is Associated with Severe and Early Onset of Dilated Cardiomyopathy" Genes 12, no. 6: 883. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060883